2.1 PRINCIPALES ASPECTOS Y NOVEDADES
El Reglamento Europeo de Productos Sanitarios (MDR 2017/745), Reglamento (UE) 2017/745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios, por el que se modifican la Directiva 2001/83/CE, el Reglamento (CE) nº178/2002 y el Reglamento (CE) nº1223/2009 y por el que se derogan las Directivas 90/385/CEE y 93/42/CEE del Consejo, representa un marco legal más estricto y exhaustivo para la regulación de dispositivos médicos en la Unión Europea. se desarrolla en 15 capítulos y XVII anexos donde se expone la normativa que regula la fabricación, la introducción al mercado y puesta en servicio permitiendo la libre circulación y comercialización y el uso de productos sanitarios en la Unión Europea (UE) con el objetivo de garantizar un alto nivel de seguridad y protección de la salud para los pacientes y usuarios apoyando la innovación.
Para los productos de diagnóstico in vitro aplica el Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
Existen otros Reglamentos posteriores que modifican las disposiciones transitorias de ambos reglamentos ampliando los plazos de implantación. Estos son:
Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022 por el que se modifica el Reglamento (UE) 2017/746 en lo que respecta a las disposiciones transitorias para determinados productos sanitarios para diagnóstico in vitro y a la aplicación diferida de las condiciones aplicables a los productos fabricados y utilizados exclusivamente en centros sanitarios
Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023 por el que se modifican los Reglamentos (UE) 2017/745 y (UE) 2017/746 en lo que respecta a las disposiciones transitorias relativas a determinados productos sanitarios y a productos sanitarios para diagnóstico in vitro
Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024, por el que se modifican los Reglamentos (UE) 2017/745 y (UE) 2017/746 en lo que respecta a la aplicación gradual de Eudamed, la obligación de informar en caso de interrupción o cese en el suministro y las disposiciones transitorias para determinados productos sanitarios para diagnóstico in vitro, amplía los periodos transitorios del Reglamento 2017/746 para garantizar la disponibilidad de productos de diagnóstico in vitro de alto riesgo. Permitirá la aplicación gradual de EUDAMED por módulos, lo que mejorará la transparencia y proporcionará información sobre los productos en el mercado de la UE. Incluye un nuevo requisito a fabricantes: informar en caso de interrupción o cese en el suministro de productos sanitarios y productos de diagnóstico in vitro esenciales
En las consideraciones del reglamento, nos marcan sus objetivos, por un lado, se quiere garantizar el buen funcionamiento del mercado de los productos sanitarios, protegiendo a pacientes y usuarios y considerando los intereses de las pequeñas y medianas empresas que desarrollan la actividad en el sector y fijando normas elevadas de calidad y seguridad para responder a las preocupaciones de seguridad planteadas.
Posteriormente se revisarán los aspectos relevantes y principales novedades del MDR 2017/745 que se desarrollarán y la evaluación clínica y seguimiento de postcomercialiazacion.
2.1.1 Ampliación del Alcance
El MDR amplía su ámbito de aplicación para incluir productos que no tienen una finalidad médica pero que se comercializan como si la tuvieran, como equipos para cirugía estética, equipos utilizados para reducir, retiras o destruir tejido adiposo para dispositivos médicos, y otros que hasta ahora no estaban regulados según lo recoge en el Capítulo I, articulo 1.
“El presente Reglamento establece normas relativas a la introducción en el mercado, la comercialización o la puesta en servicio de productos sanitarios para uso humano y accesorios de dichos productos en la Unión. El presente Reglamento también se aplica a las investigaciones clínicas relativas a los productos sanitarios y accesorios que se llevan a cabo en la Unión. 2.
El presente Reglamento será también aplicable, a partir de la fecha de aplicación de las especificaciones comunes adoptadas con arreglo al artículo 9, a los grupos de productos que no persiguen fines médicos enumerados en el anexo XVI, teniendo en cuenta los conocimientos más recientes de la medicina y, en particular, las normas armonizadas existentes para productos análogos con fines médicos, basados en una tecnología similar.
Las especificaciones comunes relativas a cada uno de los grupos de productos enumerados en el anexo XVI se referirán, al menos, a la aplicación de la gestión del riesgo expuesta en el anexo I respecto del grupo de productos en cuestión y, cuando sea necesario, a la evaluación clínica relativa a la seguridad. Las especificaciones comunes necesarias se adoptarán a más tardar el 26 de mayo de 2020. Serán aplicables a partir de seis meses después de la fecha de su entrada en vigor o a partir del 26 de mayo de 2020, si esta fecha es posterior.
No obstante, lo dispuesto en el artículo 122, las medidas de los Estados miembros por lo que respecta a la calificación de los productos a que se refiere el anexo XVI como productos sanitarios con arreglo a la Directiva 93/42/CEE seguirán siendo válidas hasta la fecha de aplicación mencionada en el párrafo primero de las especificaciones comunes pertinentes para este grupo de productos.
El presente Reglamento también se aplica a las investigaciones clínicas llevadas a cabo en la Unión relativas a los productos a que se refiere el párrafo primero…..”
La regulación es más estricta sobre los programas informáticos, software médico, que se consideran dispositivos médicos, incluyendo aquellos utilizados para el diagnóstico o tratamiento.
2.1.2 Clasificación de Productos
El MDR introduce cambios en la clasificación de los productos sanitarios, particularmente en lo que respecta a los productos de nuevos materiales, a los productos combinados, software médico y dispositivos reutilizables.
Con respecto a los procedimientos para la fabricación de productos para su uso en el propio centro sanitario y para el reprocesamiento de productos de un solo uso y su utilización, lo deja un poco abierto a que cada Estado miembro lo regule, en España queda recogido en el RRD 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
Los productos que ya están en el mercado deben ser reevaluados bajo los nuevos criterios en los plazos establecidos, lo que podría cambiar su clasificación y los requisitos asociados a los mismos.
2.1.3 Requisitos más estrictos para la Evaluación de Conformidad. Organismos notificados
El reglamento otorga un peso mayor a los organismos notificados dedicando un capítulo completo y un anexo. En él nos habla de sus requisitos que son más rigurosos, de las auditorias que deben someterse y como desarrollar sus funciones con respecto a la evaluación de la conformidad.
Se exige una documentación técnica más completa y detallada, debe incluir información sobre el diseño y fabricación, pruebas de seguridad y funcionamiento, estudios realizados, evaluación del riesgo y análisis de la relación beneficio-riesgo.
2.1.4 Sistema EUDAMED y Transparencia
Base de Datos EUDAMED: Se establece una Base de Datos Europea sobre Productos Sanitario, EUDAMED. Esta base de datos incluye información sobre el registro de productos, vigilancia, certificaciones y seguimiento clínico, y es accesible tanto para las autoridades como para el público, se pretende mejorar la transparencia y trazabilidad de los dispositivos médicos. Además, incluye un sistema de identificación única (UDI) de los dispositivos a lo largo de toda la cadena de suministro, lo que facilita el seguimiento y la gestión de productos defectuosos o retirados del mercado. Actualmente está en desarrollo.
2.1.5 Evaluación Clínica y Seguimiento Post-comercialización.
Requisitos de evaluación clínica: Se requiere una evaluación más robusta para demostrar la seguridad y eficacia del producto, incluye la obligación de realizar estudios y presentar la documentación de estos.
Vigilancia post-comercialización (PMS): El MDR introduce un sistema más riguroso de vigilancia post-comercialización, que obliga a los fabricantes a monitorizar durante toda la vida del producto el desempeño de sus productos y a informar sobre incidentes adversos y acciones correctivas.
2.1.6 Mayor Enfoque en la Seguridad y Gestión de Riesgos
Requisitos de Seguridad y Desempeño: Se refuerzan los requisitos de seguridad y desempeño, requiriendo que todos los productos cumplan con estándares más altos antes de ser comercializados lo que incluye una mayor atención a la biocompatibilidad, ciberseguridad y riesgos asociados al uso de dispositivos.
Los fabricantes deben tener un enfoque integral del producto valorándolos tanto para la gestión del riesgo como para mantener la vigilancia activa de los productos con su ciclo de vida.
2.1.7 Responsabilidades Claras para los Actores del Mercado
El reglamento clarifica y amplía las responsabilidades de todos los implicados en el ciclo de vida del producto fabricantes, representantes autorizados, importadores y distribuidores.
2.2 DEFINICIÓN DE PRODUCTO SANITARIO.
Los nuevos Reglamentos definen los productos sanitarios como:
“Producto Sanitario es cualquier instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo, destinado por el fabricante a ser utilizado en personas, por separado o en combinación, con alguno de los siguientes fines médicos específicos:
- Diagnóstico, prevención, seguimiento, predicción, pronóstico, tratamiento o alivio de una enfermedad.
- Diagnóstico, seguimiento, tratamiento, alivio o compensación de una lesión o de una discapacidad.
- Investigación, sustitución o modificación de la anatomía o de un proceso o estado fisiológico o patológico,
- Obtención de información mediante el examen in vitro de muestras procedentes del cuerpo humano, incluyendo donaciones de órganos, sangre y tejidos. Y que no ejerza la acción principal prevista en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.”
También se consideran productos sanitarios:
a) Los productos de control o apoyo a la concepción
b) Los destinados específicamente a la limpieza, desinfección o esterilización de otros productos sanitarios.”
Con esta definición podemos concluir que el producto sanitario se destina a ser utilizado con una finalidad sanitaria que vendrá determinada por la etiqueta y las instrucciones de uso establecidas por el fabricante.
Dentro de los productos sanitarios podemos encontrar:
“Producto implantable: es todo producto, incluidos los que son absorbidos parcial o totalmente, que se destina a:
- Ser introducido totalmente en el cuerpo humano o
- Sustituir una superficie epitelial o la superficie ocular, mediante intervención médica, y a permanecer en su lugar después de la intervención.
Se considerará asimismo producto implantable todo producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días; P implantable activo”
“Producto sanitario para diagnóstico in vitro: cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, kit, instrumento, aparato, pieza de equipo, programa informático o sistema, utilizado solo o en combinación, destinado por el fabricante a ser utilizado in vitro para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, única o principalmente con el fin de proporcionar información sobre uno o varios de los elementos siguientes:
- relativa a un proceso o estado fisiológico o patológico;
- relativa a deficiencias físicas o mentales congénitas;
- relativa a la predisposición a una dolencia o enfermedad;
- para determinar la seguridad y compatibilidad con posibles receptores;
- para predecir la respuesta o reacción al tratamiento;
- para establecer o supervisar las medidas terapéuticas.
Los recipientes para muestras se considerarán también productos sanitarios para diagnóstico in vitro”
Debemos recordar que la normativa de los productos sanitarios para diagnostico in vitro está recogida en el Reglamento 746/2017 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro.
Producto a medida: todo producto fabricado especialmente según la prescripción médica de cualquier persona autorizada por la legislación nacional en virtud de su cualificación profesional, en la que constan, bajo la responsabilidad de dicha persona, las características específicas de diseño, y que está destinado a ser utilizado únicamente por un paciente determinado con el fin exclusivo de atender a su estado y necesidades particulares.
Producto sanitario activo: todo producto cuyo funcionamiento depende de una fuente de energía distinta de la generada por el cuerpo humano a este efecto o por la gravedad, y que actúa cambiando la densidad de esta energía o convirtiendo esta energía. No se considerarán productos activos los productos destinados a transmitir energía, sustancias u otros elementos entre un producto activo y el paciente, sin ningún cambio significativo. Un programa informático también se considerará un producto activo.
Producto destinado a la investigación clínica el producto destinado a ser puesto a disposición del personal sanitario para llevar a cabo las investigaciones clínicas según los requerimientos generales en relación a las investigaciones clínicas con productos sanitarios en el capítulo VI y los anexos XIV y XV establecidas en el Reglamento.
También nos define los accesorios de un producto sanitario como un artículo que, sin ser en sí mismo un producto sanitario, está destinado por su fabricante a ser usado de forma conjunta con uno o varios de dichos productos, para permitir específicamente que el producto o productos sanitarios puedan utilizarse con arreglo a su finalidad prevista o para contribuir específica y directamente a la funcionalidad médica de los productos sanitarios a efectos de su finalidad prevista.
2.3 CLASIFICACIÓN DE LOS PRODUCTOS SANITARIOS POR LA UTILIDAD DE LOS CENTROS Y CATALOGACION
La clasificación de los productos sanitarios podríamos definirla de tres maneras:
- Por la utilización en los centros
- Por la catalogación según tipo de producto
- Por la clasificación de los productos según el Reglamento, clases de riesgo.
La clasificación del material por la utilización en los centros se debería considerar una clasificación por su uso que nos ayuda a entender la forma de funcionamiento de compras, almacén y reposición de las unidades, por ello se incluye material que se utiliza en los centros que no son productos sanitarios como tal.
Material fungible de un solo uso. Aquel que se consume durante su utilización o que debe ser desechado tras su uso, independientemente de su precio (actualmente en quirófanos, hemodinamia, radiología vascular se utilizan productos fungibles de alto valor económico). Por ello, quedan excluidos del inventario general de bienes y derechos. Este es un material de uso corriente en todo centro sanitario y su adquisición se realiza a lo largo de todo el año. Ejemplos; además del material de quirófanos, hemodinámica y radiología vascular, se incluye material de uso habitual en las unidades como gasas, sistemas de infusión, jeringas, etc.
Material Reutilizable. Material que se utiliza de forma repetida y necesita reprocesamiento como lavado, desinfección y/o esterilización para poder ser utilizado en otro paciente. Ejemplos pueden ser tubos de endoscopio, cajas de instrumental quirrgico
Material inventariable. Material que va a permanecer en la entidad que lo adquiere y que no va dirigido a su venta. Tiene un periodo largo de uso, utilizándose en muchas ocasiones y generalmente tiene un coste elevado y necesita una valoración más profunda para su adquisición. Dentro de este grupo están el material clínico, material quirúrgico, mobiliario clínico. Según el material de que se trate tendrá una vida útil diferente, este periodo debe ser tal que asegure una buena amortización del producto. Su adquisición se realiza en el plan de inversiones anual o en planes específicos de la comunidad autónoma. Algunos ejemplos son: TAC, electrocardiógrafo, desfibrilador, pie de goteo, cama de paciente, mesilla de noche, mesa quirúrgica.
Pequeño material. Es material de oficina, repuestos, material clínico y de laboratorio de corta duración. Todo aquel que no permanece más de un ejercicio económico, así como la papelería, libros y revistas (estos últimos se inventarían en el catálogo de biblioteca y se asigna su adquisición a un ejercicio económico). Se usan en muchas ocasiones, el precio es bajo y se adquiere en conjunto o por separado. Es un grupo que se situaría en medio de los dos grandes grupos descritos. Se refiere a material no inventariable pero que puede ser reutilizado muchas veces. Su adquisición, normalmente, se realiza en el plan de inversiones anual. Por ejemplo; aparatos de tensión arterial, pinzas de curas, sujeciones mecánicas.
La clasificación por la catalogación del producto está recogida en la norma UNE-EN ISO 15225:2016 Productos sanitarios. Gestión de la calidad. Estructura de los datos de nomenclatura para productos sanitarios. En ella se establecen los grupos genéricos para establecer la nomenclatura y facilitar el intercambio de información.
- Productos Sanitarios Implantables Activos.
- Productos para Anestesia y Respiración.
- Productos Dentales.
- Productos Electromédicos/Mecánicos.
- Equipamiento Hospitalario.
- Productos Sanitarios para Diagnóstico “In Vitro”.
- Productos Sanitarios Implantables No Activos.
- Productos Oftálmicos y Ópticos.
- Instrumentos Reutilizables.
- Productos de un Solo Uso.
- Ayudas Técnicas para Discapacitados.
- Productos que Utilizan Radiación para Diagnóstico y Terapéutica.
El primer catálogo que se realizó en España fue el del antiguo INSALUD, actualmente las Comunidades Autónomas cuentan con un catálogo centralizado propio donde tienen clasificado todo el material de los centros de forma común para poder compartir material más fácilmente en caso de necesidad entre sus centros o poder realizar adquisiciones agrupadas.
2.4 CLASIFICACION SEGUN REGLAMENTO (UE) 2017/745. REGLAS DE CLASIFICACION
La clasificación según el Reglamento (UE)2017/745 divide a los productos en las clases I, IIa, IIb y III, teniendo en cuenta la finalidad prevista de los productos, sus riesgos inherentes, el tiempo de uso y la forma de uso. La clasificación se hará según el anexo VIII, donde se desarrollan los diferentes criterios de clasificación, para ello debemos conocer ciertas definiciones y normas, con relación a las reglas de clasificación.
En caso de controversia entre el fabricante y el organismo notificado por razón de la aplicación de los criterios de clasificación, se remitirá para decisión a las autoridades competentes de las que dependa dicho organismo. En caso de tratarse de un organismo notificado español, se acudirá a la Agencia Española de Medicamentos y Productos Sanitarios.
La Agencia Española de Medicamentos y Productos Sanitarios decidirá sobre la clasificación que corresponde a los productos aplicando los criterios establecidos y podrá presentar a la Comisión Europea una solicitud cuando las reglas de clasificación requieran de una decisión para un determinado producto o para una excepción
Definiciones para tener en cuanta de cara a la clasificación de los productos
Por su duración
- Uso pasajero: destinados a utilizarse de forma continua durante menos de 60 minutos.
- Corto plazo: destinados normalmente a un uso continuado desde 60 minutos hasta 30 días.
- Uso prolongado: destinados normalmente a un uso continuado de más de 30 días.
Por su forma de uso
- Producto no invasivo.
- Producto invasivo: producto que penetra completa o parcialmente en el interior del cuerpo, bien por un orificio corporal o a través de la superficie del cuerpo. Por ejemplo, un catéter o una sonda nasogástrica
- Producto invasivo de tipo quirúrgico: Producto invasivo que penetra en el interior del cuerpo a través de la superficie corporal por medio de una intervención quirúrgica o en el contexto de una intervención quirúrgica.
- Producto implantable: producto, incluidos los que son absorbidos parcial o totalmente, que se destina a ser introducido totalmente en el cuerpo humano, o para sustituir una superficie epitelial o la superficie ocular, mediante intervención médica y a permanecer en su lugar después de la intervención. Se considerará asimismo producto implantable cualquier producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días.
Otras definiciones:
- Instrumento quirúrgico reutilizable: instrumento destinado a fines quirúrgicos para cortar, perforar, serrar, escarificar, raspar, pinzar, retraer, recortar o procedimientos similares, sin estar conectado a un producto sanitario activo, y destinado por el fabricante a ser reutilizado una vez efectuados los procedimientos adecuados tales como limpieza, desinfección y esterilización.
- Producto sanitario activo: humano a este efecto o por la gravedad, y que actúa cambiando la densidad de esta energía o convirtiendo esta energía. No se considerarán productos activos los productos destinados a transmitir energía, sustancias u otros elementos entre un producto activo y el paciente, sin ningún cambio significativo. Un programa informático también se considerará un producto activo.
- Producto activo terapéutico: cualquier producto activo, utilizado solo o en combinación con otros productos, destinado a sostener, modificar, sustituir o restaurar funciones o estructuras biológicas en el contexto del tratamiento o alivio de una enfermedad, lesión o deficiencia.
- Producto activo para diagnóstico y vigilancia: cualquier producto activo, utilizado solo o en combinación con otros productos, destinado a proporcionar información para la detección, el diagnóstico, la observación o el tratamiento de estados fisiológicos, de estados de salud, de enfermedades o de malformaciones congénitas
- Sistema circulatorio central: En el marco del Reglamento se entenderá los vasos sanguíneos siguientes: arteriae pulmonales, aorta ascendens, arcus aortae, aorta descendens hasta la bifurcatio aortae, arteriae coronariae, arteria carotis communis, arteria carotis externa, arteria carotis interna, arteriae cerebrales, truncus brachiocephalicus, venae cordis, venae pulmonales, vena cava superior y vena cava inferior.
- Sistema nervioso central: En el marco del Reglamento se entenderá “sistema nervioso central”, el cerebro, las meninges y la médula espinal.
Normas de desarrollo para la clasificación
La aplicación de las reglas de clasificación se regirá por la finalidad prevista de los productos.
Si un producto en cuestión se destina a utilizarse en combinación con otro producto, las reglas de clasificación se aplicarán a cada uno de los productos por separado. Los accesorios para un producto sanitario y para un producto enumerado en el anexo XVI serán clasificados por sí mismos por separado del producto con el que se utilicen.
Los programas informáticos que sirvan para manejar un producto o tengan influencia en su utilización se incluirán en la misma clase que el producto. Si el programa informático es independiente de cualquier otro producto, será clasificado por sí mismo.
Si un producto no se destina a utilizarse exclusiva o principalmente en una parte específica del cuerpo, se considerará para la clasificación su utilización especificada más crítica.
Si para el mismo producto son aplicables varias reglas o si, dentro de la misma regla, son aplicables varias subreglas, teniendo en cuenta la finalidad prevista del producto, se aplicarán la regla y subregla más estricta que dé lugar a la clasificación más elevada.
Por utilización continua se entenderá:
- Toda la duración de uso del mismo producto, sin importar la interrupción temporal de su uso durante un procedimiento o la retirada temporal con fines de limpieza o desinfección del producto. El carácter temporal de la interrupción del uso o la retirada se establecerá en relación con la duración del uso antes y después del período en el que el uso se interrumpe o el producto se retira, y
- El uso acumulado de un producto destinado por el fabricante a ser sustituido inmediatamente por otro del mismo tipo. 3.7. Se considerará que un producto permite un diagnóstico directo cuando proporciona el diagnóstico de la enfermedad o la afección en cuestión por sí mismo o cuando proporciona información decisiva para el diagnóstico.
Se considerará que un producto permite un diagnóstico directo cuando proporciona el diagnóstico de la enfermedad o la afección en cuestión por sí mismo o cuando proporciona información decisiva para el diagnóstico.
Las reglas de clasificación están recogidas en el Reglamento en anexo VIII, capitulo III. Se desarrollan clasificadas en productos no invasivos, invasivos, activos y reglas especiales.
Una vez conocidas las definiciones, normas y reglas de clasificación, podemos saber que según los riesgos potenciales que pueden derivarse de la utilización de los productos sanitarios y aplicando las reglas de decisión que se basan en la vulnerabilidad del cuerpo humano, los productos sanitarios se clasifican siguiendo lo que indica el Reglamento (UE) 2017/745
En general los productos de clase I son productos que corresponden a procedimientos con menor riesgo. Según aumenta la clasificación aumenta el riesgo, por ello, los de clase III se corresponden con los de mayor riesgo para el paciente.
Se puede indicar a modo orientativo y no exhaustivo debido a la complejidad de los criterios y reglas de clasificación que se encuentran en los textos, la siguiente clasificación:
Clase I
- Productos que no entran en contacto con el paciente o que entran en contacto solo con la piel intacta.
- Productos que penetran por un orificio corporal como la boca o nariz, de uso pasajero. A corto plazo los introducidos por cavidad oral hasta faringe, conducto auditivo externo hasta tímpano y cavidad nasal
- Productos destinados a ser utilizados como barrera mecánica, compresión o absorción de exudados.
Se excluyen de esta clase los productos que, aunque no entran en contacto con el paciente, pueden influir en procesos fisiológicos (productos que tratan la sangre destinada a reinfundirse) o los que suministran energía al cuerpo humano (equipos de radiodiagnóstico).
Ejemplos de productos sanitarios de esta clase son: bolsas de orina, vendas, medias elásticas, andadores, bastones y enemas.
En esta clase se incluyen también las siguientes subclases:
Clase I estériles
Son de la clase I con presentación estéril.
Ejemplos de estos son: guantes de examen estériles, jeringuillas, gasas estériles para proteger heridas Y equipos de infusión por gravedad
Clase I con función de medición
Son de la clase I y sirven para medir.
Ejemplos de estos son: tonómetros o termómetros no electrónicos.
Clase IIa
- Productos que se introducen por orificio corporal o por medios quirúrgicos, a través de la piel, y no están destinados a permanecer en él.
- Productos no invasivos destinados a la conducción o almacenamiento de sangre, fluidos, o tejidos corporales, líquidos o gases destinados a una perfusión, administración introducción en el cuerpo humano
- Productos que modifican procesos fisiológicos siempre que no se efectué de forma potencialmente peligrosa.
- Productos que se utilizan el tratamiento de heridas no incluidos en las clases I y IIb.
- Productos que se introducen por orificio corporal o por medios quirúrgicos, a través de la piel, de uso a corto plazo. De uso prolongado los introducidos por cavidad oral hasta faringe, oído externo hasta tímpano y cavidad nasal que no pueden sr absorbidos por la membrana mucosa.
- Productos invasivos en orificios corporales, salvo los quirúrgicos, destinados a conectarse a producto sanitario activo de clase IIa o superior.
- De forma general los productos invasivos de tipo quirúrgico destinados a uso pasajero o corto plazo, excepto los indicados en la clase IIb y III
- Productos implantables y productos invasivos que se coloquen dentro de los dientes.
- Desinfectantes de productos no invasivos.
Ejemplos de productos de la clase IIa son: circuitos de circulación extracorpórea, sondas urológicas, drenajes quirúrgicos, agujas, catéteres, guantes quirúrgicos, lentes de contacto y esfigmomanómetros.
Clase IIb
- Productos no invasivos que se utilizan para influenciar los procesos fisiológicos o que administran sustancias o energía de forma potencialmente peligrosa y los destinados al diagnóstico de las funciones vitales.
- Productos no invasivos destinados a modificar la composición biológica o química de células o tejidos humanos, de la sangre, de otros líquidos corporales o de otros líquidos destinados a implantarse o administrarse en el cuerpo, salvo si el tratamiento para el que el producto se usa consiste en filtración, centrifugación o intercambios de gases o de calor.
- Productos no invasivos que se utilizan para las heridas con ruptura de la dermis y sólo pueden cicatrizar por segunda intención.
- Productos que se introducen por orificio corporal o por medios quirúrgicos, a través de la piel, de uso prolongado.
- Productos invasivos que se destinen para administrar energía al cuerpo humano o intercambiarla con el mismo de forma potencialmente peligrosa, teniendo en cuenta la naturaleza, la densidad y el punto de aplicación de la energía
- Todos los productos activos destinados a emitir radiaciones ionizantes con fines terapéuticos, incluidos los productos para controlar o supervisar dichos productos, o que influyan directamente en el funcionamiento de los mismos.
- Productos invasivos de uso pasajero para ejercer un efecto biológico o a ser absorbidas totalmente o en gran parte.
- Productos anticonceptivos o para prevención de enfermedades de transmisión sexual.
- Productos desinfectantes para productos invasivos y los de limpieza de lentes de contacto.
- Productos invasivos de uso a corto plazo excepto los que pertenecen a la clase IIa.
- Productos invasivos de uso prolongado plazo excepto los que pertenecen a la clase IIa y III.
- Productos implantables que no sean IIa o III.
Ejemplos de productos de la clase IIb son: lentes intraoculares, suturas quirúrgicas no absorbibles, apósitos para heridas que cicatrizan por segunda intención, bolsas de sangre, plumas de insulina, equipos de RX diagnósticos, máquinas de anestesia y sistemas de vigilancia de cuidados intensivos.
Clase III
- Productos invasivos destinados a entrar en contacto con el sistema nervioso central y el sistema circulatorio central para diagnóstico, control y tratamiento.
- Productos que se absorben totalmente y los que contienen sustancias medicinales.
- Los implantes mamarios y las prótesis articulares de cadera, rodilla y hombro
Ejemplos de productos de la clase III son: suturas absorbibles, adhesivos internos biológicos, endoprótesis vasculares (stents), prótesis de cadera, válvulas cardiacas y catéteres recubiertos de medicación.
Además de esta clasificación de los productos sanitarios encontramos los productos a medida realizados para un paciente en particular, productos para investigaciones clínicas y los productos para diagnostico in vitro.
2.5 ETIQUETADO DE PRODUCTOS SANITARIOS. INSTRUCCIONES DE USO
Todos los productos sanitarios deben estar identificados según indica el RD con la información necesaria para su utilización con seguridad. El fabricante indicará en cada producto la información necesaria para su utilización con plena seguridad, teniendo en cuenta la formación y los conocimientos de los usuarios potenciales y para identificar al fabricante. La información constará de las indicaciones que figuren en la etiqueta y las que figuren en las instrucciones de utilización.
Los datos necesarios para la utilización del producto deberán figurar, cuando sea factible, en el propio producto y/o en un envase unitario o en el envase comercial. Si no es factible envasar individualmente cada unidad, estos datos deberán figurar en unas instrucciones de utilización que acompañen a uno o varios productos. Excepcionalmente, las instrucciones no serán necesarias en el caso de los productos de las clases I y IIa.
El marcado de cumplimiento de RD es CE con unas dimensiones establecidas, si el marcado debe disminuirse o aumentarse se realizará en proporción. Existen una serie de datos que deben aparecer siempre en las etiquetas de los productos, cuando sea apropiado podrán ser en forma de símbolo deben ajustarse a las normas armonizadas. Siempre debe incluir:
- Nombre del fabricante y distribuidor autorizado si el fabricante no tiene domicilio social en la Unión Europea.
- Información para identificación del producto y contenido del envase.
- Si es estéril debe aparecer indicando método de esterilización.
- El número de lote de fabricación o de serie.
- Fecha de caducidad expresada con mes y año. En los productos activos el año de fabricación.
- Si es de un solo uso.
- En los productos a medida la indicación de ello “producto a medida”.
- Si es un producto solo para investigación clínica debe quedar reflejado “exclusivamente para investigaciones clínicas”.
- Condiciones de almacenamiento y conservación.
- Instrucciones especiales de utilización.
- Cualquier advertencia o precaución que deba adoptarse. Por ejemplo, si tiene látex.
- Método de esterilización, si es preciso
- Indicación de que el producto contiene como parte integrante una sustancia derivada de la sangre humana, si es preciso.
- Marcado CE con las medidas establecidas en el RD, si cambia de tamaño deberá mantener la proporción. El requisito de dimensiones se puede no cumplir en caso de productos muy pequeños.
- Sistema UDI.
Las instrucciones uso de forma general deben incluir los datos suficientes para la comprensión por el usuario:
- Los datos del etiquetado.
- La forma de utilización.
- Los posibles efectos secundarios.
- Precauciones a tomar en su utilización.
- Instrucciones de montaje (si es necesario).
- Mantenimiento (si es necesario).
- La fecha de publicación de la última revisión de las instrucciones.
Además, si es un producto reutilizable deberá aparecer los procedimientos de uso, limpieza, desinfección y esterilización, si es un producto sanitario de solo uso, los riesgos que supondría reutilizarlo y si es un producto con función de medición, el grado de precisión.
Los datos relacionados con los productos sanitarios pueden adoptar, cuando sea apropiado, la forma de símbolos, pictogramas. Los símbolos y los colores de identificación que se utilicen deberán ajustarse a las normas armonizadas. Si no existe ninguna norma al respecto, los símbolos y los colores se describirán en la documentación que acompañe al producto. En el caso del etiquetado del producto sanitario la norma existente es la UNE-EN ISO 15223-1:2022 Productos sanitarios. Símbolos a utilizar con la información a suministrar por el fabricante. Los símbolos ajustados a normas armonizadas más utilizados para el etiquetado son:
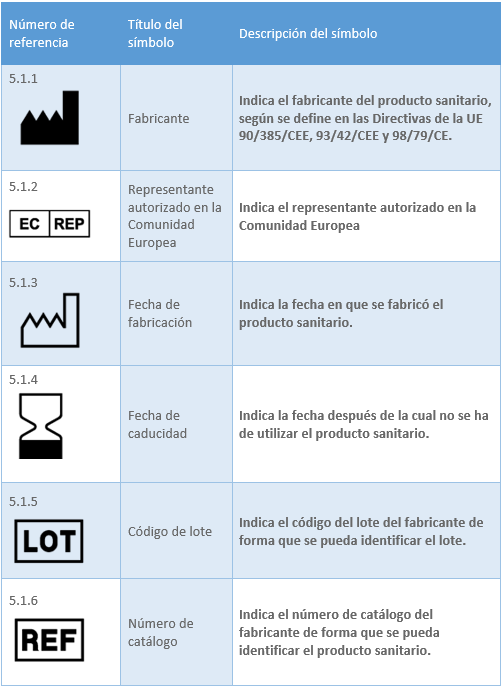
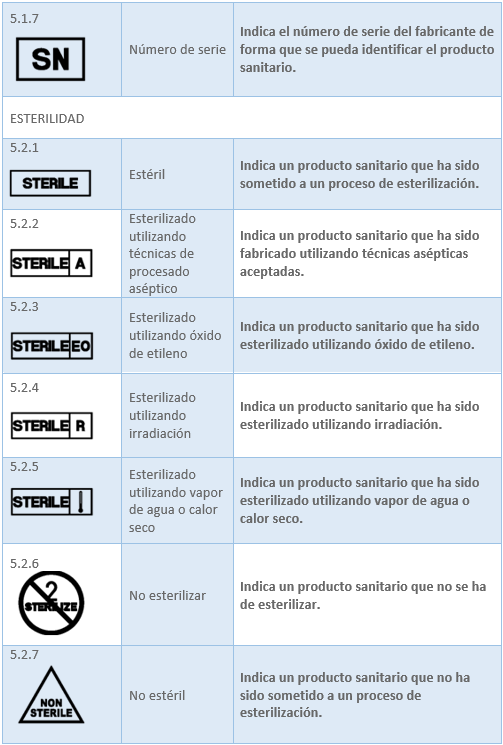
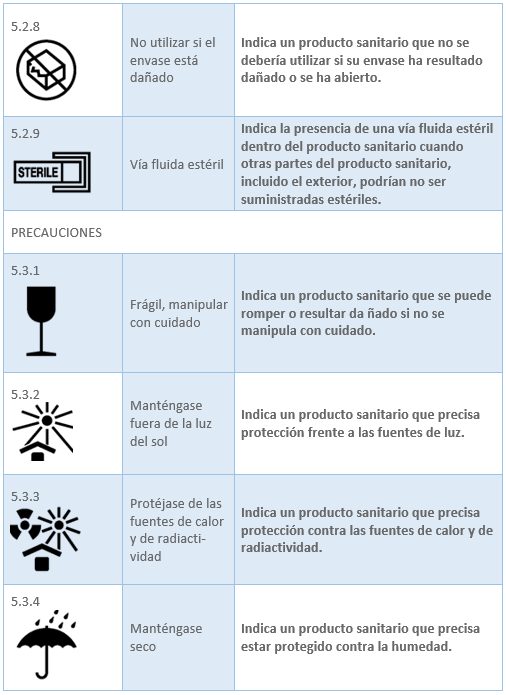
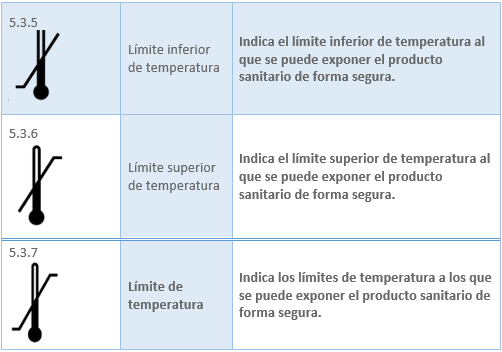
2.6 MARCADO CE. ORGANISMO NOTIFICADO. REQUISITOS Y PROCESO DE OBTENCIÓN DEL MARCADO CE
El Marcado CE es el símbolo que indica que el producto sanitario cumple con los requisitos de seguridad y eficacia establecidos por la legislación europea. Es un requisito obligatorio para la comercialización y libre circulación de productos sanitarios en Europa. Desde 1998, ningún producto sanitario puede comercializarse en los países europeo sin este marcado según la Directiva 93/42/CEE, vigente hasta la entrada en vigor del Reglamento (UE)2017/745. Se coloca tras un procedimiento de evaluación de la conformidad, y es obligatorio para todos los productos sanitarios comercializados en la UE. Tiene un periodo de validez de 5 años y es prorrogable de 5 en 5 años.
Se indica en su etiquetado e instrucciones de uso mediante el símbolo:

La identificación sin número es la autocertificación y con número se debe a que se realiza por un Organismo Notificado. Cada Organismo Notificado tiene un número distintivo. La clasificación del producto (clases I, IIa, IIb y III) determina los procedimientos de evaluación de la conformidad y la implicación de los organismos notificados (excepto para los productos sanitarios de Clase I). Los productos de mayor riesgo (clases IIb y III) están sujetos a controles más rigurosos, como paneles de expertos y ensayos clínicos.
Para poder venderse en el Espacio Económico Europeo, o por sus siglas, el EEE (formado por la UE más Islandia, Liechtenstein y Noruega), los productos deben llevar obligatoriamente el marcado CE, que constituye la prueba de que el producto se ha evaluado y cumple los requisitos de seguridad, sanidad y protección del medio ambiente exigidos por la UE. Es válido para los productos fabricados tanto dentro como fuera del EEE, cuya comercialización esté prevista dentro del mismo. A través del siguiente link se puede comprobar el índice de productos con distintivo CE; http://europa.eu/youreurope/business/product/ce-mark/index_es.htm
Por último, cabe señalar que el cumplimiento de estos requisitos no solo garantiza la seguridad del paciente, sino que también refuerza la transparencia y la competitividad del sector. La planificación cuidadosa del proceso de comercialización, teniendo en cuenta la normativa vigente y las directrices de la AEMPS, es esencial para el éxito de los productos sanitarios en el mercado español.
Un Organismo Notificado es una organización designada por un Estado miembro de la Unión Europea para evaluar la conformidad de determinados productos antes de su comercialización. Son los que emiten los certificados correspondientes, siguiendo los procedimientos establecidos para la evaluación de los productos autorizando la comercialización y realizan el seguimiento de la postcomercializacion. Estos certificados tienen que mencionar alguno de los Anexos de la Directiva que corresponda (Directiva 90/385/CEE, Directiva 93/42/CEE o Directiva 98/79/CE), vigentes hasta la entrada en vigor del Reglamento (UE) 2017/745 y Reglamento (UE) 2017/746 y RD 192/2023.
La Comisión Europea creara una base de datos de organismos notificados en virtud del Reglamento donde los estados miembros introducirán los datos de los organismos para su aprobación final por la comisión, esta base de datos se denomina NANDO.
En el Reglamento se desarrollan los requisitos que deben cumplir como organización, poniendo especial interés en la gestión de la calidad indicando claramente en el Anexo VII, punto 2.1 “Los organismos notificados establecerán, documentarán, aplicarán, mantendrán y explotarán un sistema de gestión de la calidad que se ajuste a la naturaleza, el ámbito y la escala de sus actividades de evaluación de la conformidad y sea capaz de apoyar y demostrar un cumplimiento coherente de los requisitos del presente Reglamento”, todo ello encaminado al control de documentación, asignación de actividad y responsabilidades al personal, evaluación y proceso de la toma de decisiones, control de registros, auditorías internas, acciones correctivas y preventivas, reclamaciones y formación continuada.
Por otro lado, aborda cómo debe ser el proceso para la evaluación de conformidad debiendo estar debidamente protocolizado y documentado de la manera más pormenorizada posible para que los fabricantes puedan conocer como entregar la solicitud, con que documentación y que estudios son necesarios para la evaluación que le van a realizar. Indica expresamente que el proceso debe ser realizado por diferentes personas no pudiendo ser la misma persona la que realice las evaluaciones y la encargada de la revisión final.
Recoge el contenido que debe tener el informe final uniforme y con los elementos mínimos que determine el Grupo de Coordinación de Productos Sanitarios, debe incluir: las etapas de evaluación que demuestre el cumplimiento del Reglamento, las conclusiones de las valoraciones de las evaluaciones clínicas con la documentación de los resultados extraídas y la decisión del organismo.
Una vez terminado ese proceso los organismos deben realizar actividades de control y supervisión para asegurar que los productos siguen cumpliendo el Reglamento y los fabricantes deben mantener informado al organismo de cualquier incidencia en ciclo de vida del producto para que estos puedan tomar las medidas oportunas.
Para la recertificación los organismos contaran también con protocolos de documentación a presentar.
En España los organismos notificados son designados por las autoridades nacionales, que ostentan la capacidad de supervisión sobre las actividades de dichos organismos, debiendo éstos facilitarles toda la información necesaria, incluida la relativa a los certificados emitidos, rechazados, suspendidos o retirados. Está recogido en la Directiva 93/42/CEE del Consejo de Europeo, traspuesta al RD 414/1996 y posteriormente al RD 1591/2009 donde se detalla el procedimiento a seguir por el organismo notificado en sus actividades de certificación de la conformidad solicitadas por los fabricantes, hasta la entrada en vigor de los nuevos Reglamentos.
La designación del Organismo Notificado (0318) fue realizada por las Autoridades Sanitarias Españolas en 1995, para los productos sanitarios y los productos sanitarios implantables activos, en diciembre del 2002, para los productos sanitarios para diagnóstico “in Vitro” y en el 2004, para los productos sanitarios que incorporan derivados de tejidos animales. Para todos ellos, el ámbito de la designación se extiende a todos los productos en cuya evaluación de conformidad debe intervenir un Organismo Notificado y para todos los procedimientos de evaluación de conformidad. El Organismo Notificado 0318 fue redesignado nuevamente en marzo de 2010.
Posteriormente, mediante el Real Decreto 1275/2011, de 16 de septiembre, se crea la Agencia estatal “Agencia Española de Medicamentos y Productos Sanitarios” (AEMPS) y se aprueba su Estatuto. Entre sus competencias está:
“Actuar como Organismo Notificado, evaluando la conformidad de los productos sanitarios, realizando las auditorías de los sistemas de calidad, certificando las normas específicas de dichos sistemas y emitiendo los certificados CE con vistas a la colocación del marcado CE en dichos productos, en los términos que establezca la designación efectuada por el Ministerio de Sanidad, Servicios Sociales e Igualdad , así como autorizar las entidades colaboradoras en la certificación de los productos sanitarios”
Actualmente, con fecha 28 de junio 2019, la AEMPS ha presentado, la solicitud al Ministerio de Sanidad, Consumo y Bienestar Social para ser designada como Organismo Notificado de acuerdo con el Reglamento 2017/745, de 5 de abril, sobre los productos sanitarios.
La intervención del organismo notificado depende de la clasificación de riesgo del producto se evalúan distintos procesos, según se indica a continuación:
- PS Clase I: No requiere la intervención del Organismo Notificado, el fabricante realiza la autocertificación de marcado CE.
- PS Clase I estériles: La intervención del Organismo Notificado se limitará únicamente a los aspectos relacionados con la obtención y mantenimiento de las condiciones de esterilidad.
- PS Clase I con función de medición: La intervención del Organismo Notificado se limitará únicamente a los aspectos relacionados con la función de medición.
- PS Clase I estériles con función de medición: La intervención del Organismo Notificado se limitará a los aspectos relacionados con la obtención y mantenimiento de la esterilidad y con la función de medición.
- PS Clase IIa: La intervención del Organismo Notificado irá encaminada a evaluar el cumplimiento de todos los requisitos esenciales aplicables al producto. Fabricación y la esterilización.
- PS Clase IIb: La intervención del Organismo Notificado irá encaminada a evaluar el cumplimiento de todos los requisitos esenciales aplicables al producto. Diseño, fabricación, esterilización y ensayos clínicos.
- PS Clase III: La intervención del Organismo Notificado irá encaminada a evaluar el cumplimiento de todos los requisitos esenciales aplicables al producto. Aprobación previa de diseño, fabricación, esterilización y ensayos clínicos
El proceso para la obtención del certificado CE es:
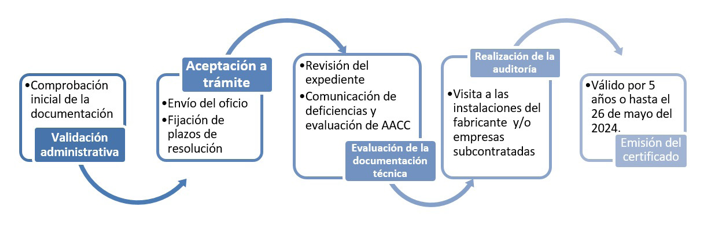
Fuente: Organismo Notificado 0318. Informacion y condiciones para el marcado CE.
La certificación de los productos sanitarios se realiza teniendo en cuenta la clasificación que como ya sabemos, puede ser Clase I, IIa, IIb o III siendo esta ultima la que supone mayor riesgo según sus características y el riesgo derivado de su uso y por ello más exigente la certificación. Está recogido en los Anexos I al XI de la MDR cómo llevarla a cabo:
- Anexo I Requisitos generales de seguridad y funcionamiento. Trata del diseño, fabricación y la información proporcionada del producto.
- Anexo II Documentación técnica. Relaciona la documentación técnica que debe elaborar el fabricante y como debe presentarse para su evaluación de forma clara, organizada, fácil de buscar e inequívoca.
- Anexo III Documentación técnica sobre poscomercializacion.
- Anexo IV Declaración UE de conformidad. Recoge toda información básica que debe contener la declaración, siendo el documento base que certifica la calidad del producto especialmente para la Clase I ya que esta clase se certifica con autocertificación.
- Anexo V Marcado CE de conformidad. Se describe como debe aparecer el CE en los productos, proporciones y tamaños mínimos. Debemos recordar que han existido logos que podían dar lugar a error, ejemplo China Export que indicaba CE pero no cumplía con las dimensiones.
- Anexo VI Informacion que debe presentarse con el registro de los productos y agentes económicos conforme al artículo 29, apartado 4 y al artículo 31, datos principales que deben facilitarse a la base de datos del sistema de identificación única junto con el UDI_DI de conformidad con los artículos 28 y 29 sistema de identificación única del producto. Los fabricantes o, en su caso, los representantes autorizados y, cuando proceda, los importadores, presentarán la información a que se refiere al agente económico y garantizarán que la información relativa a sus productos correcta y actualizada por la parte pertinente.
- Anexo VII Requisitos de los Organismos Notificados. Versa de las características de los Organismos Notificados, de la gestion de la calidad, recursos y de cómo deben documentar todos los procesos de certificación teniendo que estar suficientemente pormenorizados para la realización de cada actividad de evaluación de la conformidad para las que hayan sido designados con todas las etapas.
- Anexo VIII. Reglas de clasificación. Se explican las diferentes características del producto para su clasificación, como ya estudiamos anteriormente.
- Anexo IX. Evaluación de la conformidad basada en un sistema de gestion de la calidad y en la evaluación de la documentación técnica. Indica la necesidad de contar con un sistema de calidad donde se revise y controle la trazabilidad para garantizar lo establecido en la MDR.
- Anexo X. Evaluación de la conformidad basada en el examen de tipo. Es el procedimiento por el que un organismo notificado comprueba y certifica que un producto, con su documentación técnica y sus procesos pertinentes de ciclo de vida y una muestra representativa correspondiente de la producción, cumple las disposiciones pertinentes de este Reglamento.
- Anexo XI. Evaluación de la conformidad basada en la verificación de la conformidad del producto. El objetivo es garantizar que los productos se ajusten al tipo para el que se ha expedido un certificado de examen UE de tipo y que cumplan las disposiciones aplicables.
Los requisitos por clase de producto para la obtención de la certificación, entendiendo que son acumulativos según avanzamos en la clase de producto, son:
Común a todos los productos desde la clase I hasta la III:
- Requisitos generales de seguridad y funcionamiento. Anexo I
- Documentación técnica general y poscomercializacion. Anexos II y III
- Declaración UE de conformidad. Artículo 19.
Los productos de Clase I estériles, quirúrgicos, reutilizables o con función de medida, en los que se establecen requisitos adicionales se puede obtener en base al Anexo IX o el Anexo XI cumpliendo los criterios relativos a la calidad del producto.
Los productos de Clase IIa también existen las dos posibilidades en base a los diferentes anexos. Si se basa en el Anexo IX, se evaluará la conformidad del producto basada en el Sistema de Gestión de la Calidad del fabricante y la documentación técnica aportada ante el Organismo Notificado, solo se debe incluir un producto por categoría. Si se basa en el Anexo XI, se evalúa la conformidad basada en la verificación de la conformidad del producto, según lo estipulado en la sección 10 (calidad del proceso de producción) o conforme a la sección 18 (sistema de verificación de la producción).
Los productos de la clase IIb de nuevo existen varias posibilidades. Si se basa en el Anexo IX para productos implantables, se ha de aportar la documentación de todos los productos cubiertos por el Sistema de Gestion de Calidad y para los productos especiales se establece la realización de una evaluación adicional relacionada con el aseguramiento de la seguridad de las funciones que los distinguen del resto de productos de Clase IIb. Basándose en el Anexo XI se deben cumplir todos los requerimientos incluidos en la Parte A o la Parte B del anexo y es necesario realizar una evaluación de conformidad basada en un examen UE de tipo del producto, según el Anexo X. En este procedimiento el Organismo Notificado comprueba y certifica que un producto, incluidos su documentación técnica y sus procesos pertinentes de ciclo de vida y una muestra representativa correspondiente de la producción de este, cumple las disposiciones pertinentes del MDR.
Los productos de clase III vuelven a existir las dos vías. Según Anexo IX se ha de aportar la documentación de todos los productos cubiertos por el Sistema de Gestión de Calidad. La segunda posibilidad debe cumplir los Anexos X y XI en las disposiciones de aplicación para la clase III.
Los plazos para la obtención del marcado CE según el Reglamento se han pospuesto debido al impacto de la pandemia de Covid, en ese momento mediante el Reglamento UE 202/561 se pospuso un año quedando la aplicación en el 26 de mayo de 2021 y se mantuvo el periodo transitorio hasta el 26 de mayo 2024, que fue prorrogado posteriormente en el Reglamento UE 2022/112. A pesar del aumento constante del número de organismos notificados designados de conformidad con el Reglamento, la capacidad global era insuficiente para garantizar la evaluación del gran número de productos objeto de la certificación expedidos en conformidad a las antiguas directivas antes del 26 de mayo 2024 y por otra parte debido a los nuevos requisitos algunas empresas no están suficientemente preparadas. Además, los profesionales valoraban un riesgo real de escasez de productos por todo ello se desarrolló el Reglamento UE 2023/607 donde se variaban los plazos de la implantación y prorrogas para dar tiempo a los organismos notificados para llevar a cabo las evaluaciones de la conformidad que se exigen. Los plazos para los productos con certificado conforme a la Directiva 90/385/CEE o la Directiva 93/42/CEE y que sea válido en virtud del apartado 2 se podrán introducir en el mercado o poner en servicio hasta las siguientes fechas:
- Productos sanitarios clase III y para los productos implantables de la clase IIb, excepto material de sutura, grapas, materiales para obturación dental, aparatos de ortodoncia, coronas dentales, tornillos, cuñas, placas, cables, alfileres, clips y dispositivos de conexión - 31 de diciembre de 2027
- Productos sanitarios clase IIb distintos de los contemplados en el punto anterior del presente apartado, para los productos de la clase IIa, y para los productos de la clase I introducidos en el mercado en condiciones estériles o que tengan una función de medición – 31 de diciembre de 2028
- Los demás productos sanitarios de clase I – 31 de diciembre 2028
2.7 DOCUMENTACIÓN MÍNIMA PARA LA OBTENCIÓN DEL MARCADO CE
La documentación técnica es un conjunto completo y estructurado de información que demuestra que el producto sanitario cumple con los requisitos generales de seguridad y rendimiento del MDR. Debe ser entregada por el fabricante de forma clara, organizada, fácil de buscar y debe incluir como mínimo:
- Descripción y especificaciones del producto, incluidas las variantes donde debe constar: denominación del producto, descripción general y detallada de las piezas incluida finalidad y usos previstos, el UDI-DI básico asignado por el fabricante o cualquier otra identificación clara del producto mediante un código, pacientes destinatarios con indicaciones, contraindicaciones y advertencias, modo de acción demostrado científicamente, justificación de que es un producto sanitario y clase de riesgo según las reglas, características novedosas, descripción de los accesorios que tenga o puedan ser combinados, descripción de las diferentes variantes pensadas comercializar y descripción de las materias primas. También, si existe, se debe hacer referencia a productos de generación anterior del fabricante que sean base para este nuevo producto o de otros fabricantes que estén disponibles.
- Descripción de las etiquetas de los envases tanto unitario como de venta y transporte en las lenguas de los Estados miembro e instrucciones de uso.
- Informacion sobre el diseño y fabricación donde deben incluir las etapas del diseño, procesos de fabricación y validación e identificación de los lugares donde se llevan a cabo las diferentes actividades.
- La información sobre los requisitos generales de seguridad y funcionamiento incluidos en el Reglamento que aplican y de los que no aplican una explicación detallada de porque no, los métodos utilizados para la demostración del cumplimiento, las normas armonizadas aplicadas y la identidad exacta de los documentos controlados que demuestren la conformidad.
- Análisis de beneficio y riesgos realizados y las soluciones adoptadas a los riesgos detectados en el análisis.
- Para la verificación y validación de los productos incluirá los resultados y análisis críticos de los ensayos realizados para demostrar la conformidad del producto con los requisitos de este Reglamento y, en particular, el cumplimiento de los requisitos generales de seguridad y funcionamiento como resultados de ensayos de ingeniería, de laboratorio, de uso simulado y de uso con animales. También se deberá presentar la evaluación de la bibliografía publicada, información del diseño del ensayo, protocolos de ensayo o estudio, métodos de análisis de los datos y conclusiones, en particular en relación con la biocompatibilidad del producto, caracterización física, microbiológica y seguridad eléctrica y compatibilidad electromagnética.
Si existe programa informático también incluirá la validación del programa informático, estabilidad y vida útil.
Plan de evaluación clínica y sus actualizaciones.
Los productos estériles deben especificar el método de esterilización con los informes de validación de estos.
Los productos con función de medición una descripción de los métodos utilizados para validar la medición.
Si el producto debe conectarse con otros para su uso se debe presentar una descripción de la combinación.
En el caso de productos especiales como: los que llevan medicamentos, elaborados con células o tejidos, productos que son concebidos para ser introducidos por un orificio o aplicados en piel y compuestos por sustancias que deben ser absorbidas, productos que contengan sustancias CMR o alteradores endocrinos deben además justificarse estas circunstancias.
- Plan de seguimiento y postcomercilizacion según Reglamento. Debe incluir. como recoger y registrar la información de los incidentes y las acciones correctivas, informes de tendencia de uso e incidentes surgidos, información de productos similares, comentarios y reclamaciones de usuarios.
Este plan debe ser un proceso proactivo y sistémico con métodos eficaces y apropiados para evaluar los datos recogidos e investigar las reclamaciones, pudiendo determinar e iniciar las medidas adecuadas.
Debe recoger también como se deben comunicar con las autoridades competentes, organismos notificados, agentes económicos y usuarios.
La documentación técnica es esencial para demostrar:
- Que el producto cumple con los requisitos generales de seguridad y funcionamiento establecidos en el Anexo I.
- Que los riesgos asociados se han minimizado y el beneficio supera cualquier riesgo residual.
El fabricante debe conservar la documentación técnica durante al menos 10 años (o 15 años para los implantes) después de la última fabricación del producto.
Deberá estar disponible para las autoridades competentes y los organismos notificados cuando lo soliciten.
Declaración UE de conformidad (Anexo IV del MDR) es el documento en el que el fabricante declara formalmente que el producto cumple con todos los requisitos aplicables del MDR.
La declaración UE de conformidad debe incluir:
- Identificación clara del producto (nombre, tipo, número de serie/lote).
- Nombre y dirección del fabricante y, si corresponde, del representante autorizado.
- Referencia a los Reglamentos aplicables y normas armonizadas utilizadas.
- Nombre del organismo notificado (si ha intervenido) y número del certificado.
- Fecha de emisión y firma de la persona responsable
La declaración UE de conformidad nos permite colocar el marcado CE en el producto. Es el paso final del procedimiento de evaluación de la conformidad.
Al igual que la documentación técnica, debe conservarse durante 10 o 15 años después de la última fabricación del producto.
Información de vigilancia y seguimiento poscomercialización que debe incluir:
- Plan de vigilancia poscomercialización (PMS Plan)
- Plan de seguimiento clínico poscomercialización (PMCF Plan), si aplica.
Sistema de gestión de calidad es el requisito de calidad del fabricante, generalmente conforme a ISO 13485.
Intervención del organismo notificado que como ya sabemos según la clasificación de riesgo para productos de riesgo moderado o alto (clases IIa, IIb y III), debe incluir los certificados de conformidad emitidos por el organismo notificado que interviene, así como los de Clase I estériles, con función de medida o reutilizables en los que interviene el organismo notificado sólo en los aspectos que marcan la excepción.
2.8 EUDAMED. BASE DE DATOS EUROPEA
EUDAMED es la Base de Datos Europea sobre Productos Sanitarios creada por la Comisión Europea dividida en seis módulos. En esta base de datos se centraliza la información sobre:
- Productos sanitarios comercializados en la UE
- Fabricantes, importadores y distribuidores
- Vigilancia de mercado y certificaciones
Podrá encontrar toda la información de los PS respecto a registro, certificados, vigilancia en el mercado europeo e investigación clínica. Actualmente continua en desarrollo.
Esta información será accesible a las autoridades y al público para así mejorar la transparencia y trazabilidad de los dispositivos médicos, así se permitirá que el público este informado de los dispositivos disponibles, sus certificados e investigaciones clínicas.
La AEMPS está trabajando en la elaboración de un nuevo registro que estará integrado en EUDAMED y al que hace referencia el art. 18 del RD 192/2023 de productos sanitarios.
En la base de datos cada dispositivo estará identificado con una identificación única de producto, un número de identificación denominado UDI que lo identifica a lo largo de toda la cadena de suministro, lo que facilita el seguimiento y la gestión de productos defectuosos o retirados del mercado.
El Sistema UDI, como ya hemos comentado es uno de los grandes cambios del Reglamento de la Unión Europea 745/2017 donde aparece por primera vez. Está basado en directrices internacionales y ayuda a reducir los errores médicos y a combatir la falsificación de los productos. Debe aplicarse a todos los productos comercializados con excepción de los productos a medida, incluyendo definiciones, que sean compatibles con los utilizados por los principales socios comerciales.
UDI es un número de identificación del producto, que consta de dos partes:
- DI: parte estática es el principal identificador del modelo de producto. A su vez se divide en:
- UDI-Di Básico, es un código alfanumérico con una longitud máxima de 25 caracteres compuesta por identificación de empresa, referencia del modelo y digito de control.
- UDI-DI es la identificación de un producto sanitario como un artículo comercial identificada con formato tipo GTIN.
- PI: parte dinámica. Identifica la unidad de producción del producto. Tiene que llevar incluido el número de serie, lote, fecha de identificación y fabricación y/o caducidad.
Los formatos aprobados para la lectura de los códigos son código de barras lineal, Datamatrix o RFID, debe figurar en la etiqueta del producto y embalajes.
La parte de UDI-DI no es obligatorio que aparezca en el etiquetado, es la forma de acceder a la base de datos EUDAMED. Donde sí debe figurar es en todos los certificados y declaraciones de conformidad del producto.
La implantación del marcado UDI está recogida en el artículo 123 del Reglamento de PS con ampliación de plazo en el Reglamento (EU) 2020/561 y en el 113 en el Reglamento de PS de diagnóstico in vitro.
Los plazos de entrada en funcionamiento de los módulos de EUDAMED son:
- Registro de agentes económicos, disponible desde 2020.
- Registro de UDI/productos, disponible desde octubre 2021.
- Organismos notificados y certificados disponible desde octubre de 2021
- Investigaciones clínicas y estudios de funcionamiento en desarrollo
- Vigilancia y seguimiento postcomercialización en desarrollo
- Control de mercado en desarrollo.
En el Reglamento 2024/1860 se indica el uso obligatorio de cada módulo después de 6 meses de que se declare funcional.
Los productos sanitarios tendrán que disponer de código UDI en diferentes fechas según su clase:
- PS de Clase III y productos implantables 26 de mayo 2021
- PS Clase IIa y IIb 26 de mayo 2023
- PS Clase I 26 de mayo de 2025.
En los productos reutilizables que deban llevar sobre el propio producto el soporte de la identificación única, el artículo 27, apartado 4:
- PS de Clase III y productos implantables 26 de mayo 2023
- PS Clase IIa y IIb 26 de mayo 2025
- PS Clase I 26 de mayo de 2027.
Por último, en los productos de diagnóstico in vitro:
- Productos sanitarios de Diagnóstico in vitro de la clase D 26 de mayo 2023
- Productos sanitarios de Diagnóstico in vitro de la clase B y C 26 de mayo 2025
- Productos sanitarios de Diagnóstico in vitro de la clase A 26 de mayo 2027.
La trazabilidad de los productos mediante el Sistema UDI debe hacer que aumente significativamente la eficacia de las actividades relacionadas con la seguridad poscomercialización, al hacer posible una notificación de incidentes mejorada, unas acciones correctivas de seguridad más precisas y un mejor seguimiento por parte de las autoridades competentes.
BIBLIOGRAFÍA
- https://es.linkedin.com/pulse/el-instrumental-quir%C3%BArgico-algo-de-su-historia-albarracin-miranda.
- historia.nationalgeographic.com.es/a/sofisticacion-antiguo-egipto-protesis-hace-3000-anos_11639
- portalcomunicacion.uah.es/diario-digital/actualidad/medicina-medieval-una-etapa-oscura-pero-llena-de-supersticiones/
- www.salusplay.com/apuntes/quirofano-y-anestesia/tema-3-instrumentacion
- wfsahq.org/es/about/history/history-of-anaesthesia
- www.bbvaopenmind.com/ciencia/investigacion/joseph-lister-el-hombre-que-esterilizo-la-cirugia/
- www.revistamedicinaycultura.fmposgrado.unam.mx/index.php/2023/01/24/joseph-lister-biografia/
- identyd.com/historia-autoclave/
- www.elsevier.es/es-revista-revista-argentina-radiologia-383-articulo-wilhelm-conrad-roentgen-el-descubrimiento-S0048761916301545
- www.fda.gov/medical-devices/overview-device-regulation/history-medical-device-regulation-oversight-united-states
- formasefh.sefh.es/tecnifarmh/curso-productos-sanitarios/curso-productos-sanitarios.pdf
- www.aemps.gob.es/la-aemps/legislacion/legislacion-sobre-productos-sanitarios/
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- www.aemps.gob.es/productos-sanitarios/investigacionclinica-productossanitarios/
- UNE-EN ISO 14971:2020 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN 62366-1:2015/A1:2020 (Ratificada) Productos sanitarios. Parte 1: Aplicación de la ingeniería de usabilidad a los productos sanitarios.
- Aplicación de la gestión de riesgos a los productos sanitarios (une.org)
- 231219_GuiaValidacion_Tec_Sanitarias-1.pdf (inndromeda.es)
- Ciclo de vida de producto sanitario archivos - Consultoría de producto sanitario
- Ciclo de vida de producto sanitario y gestión de riesgos - Fernando Atienza
- Producto sanitario definición, características y usos (productossanitarios.net)
- Risk Management in the Medical Device Industry (dqsglobal.com)
- Design Thinking en Producto Sanitario ¿Qué es idear, prototipar y testar? (ambit-bst.com)
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Ley 29/2006, de 26 de julio de garantías y uso racional del medicamento.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS): https://www.aemps.gob.es/
- Federación Española de Empresas de Tecnología Sanitaria (FENIN): https://www.fenin.es/
- Ministerio de Ciencia e Innovación (PERTE Salud de Vanguardia): https://www.ciencia.gob.es/
- European Commission. (2021). Guidance on the application of the MDR and IVDR during the COVID-19 pandemic. Publications Office of the European Union. https://health.ec.europa.eu/system/files/2021-05/md_guidance_md_011_en_0.pdf
- Ghisellini, P., Cialani, C., & Ulgiati, S. (2016). A review on circular economy: The expected transition to a balanced interplay of environmental and economic systems. Journal of Cleaner Production, 114, 11-32. https://doi.org/10.1016/j.jclepro.2015.09.007
- Organización Internacional del Trabajo (OIT). (2023). Declaración de principios y derechos fundamentales en el trabajo. https://www.ilo.org/global/lang--es/index.htm
- Organización Mundial de la Salud (OMS). (2022). Health product policy and standards. https://www.who.int/teams/health-product-and-policy-standards
- Parlamento Europeo y Consejo de la Unión Europea. (2017). Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo de 5 de abril de 2017 sobre los productos sanitarios. Diario Oficial de la Unión Europea. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX%3A32017R0745
- Porter, M. E., & Kramer, M. R. (2011). Creating shared value. Harvard Business Review, 89(1/2), 62-77.
- OMS. (2015). Ética de la salud mundial: cuestiones clave.
- Declaración de Helsinki de la AMM – Principios éticos para las investigaciones médicas con participantes humanos – WMA – The World Medical Association.
- Global health ethics: key issues (who.int)
- https://lac.saludsindanio.org/cambio-climatico-y-salud/huella-climatica-del-sector-salud
- https://www.miteco.gob.es/content/dam/miteco/es/cambio-climatico/temas/mitigacion-politicas-y-medidas/docuementoexplicativosello_tcm30-479000.pdf
- https://accionclimaticaensalud.org/sites/default/files/2021-06/huellaclimatica.pdf
- Estrategia de Economía Circular de Euskadi 2030 - Prevención de la contaminación, inspección y control ambiental - Euskadi.eus
- Residuos Sanitarios (miteco.gob.es)
- Guía de procedimientos de esterilización en el medio hospitalario. file:///C:/Users/50820026P/Documents/OneDrive%20-%20Madrid%20Digital/PERSONAL/MASTER%20ordenador%20sollube/2.%20TEMARIO%20MASTER%20FARMA%20Y%20PS/procedementos_esterilizacion.pdf
- Curso básico de Gestion de Productos Sanitarios ANECORM
- https://www.aemps.gob.es/informa/la-aemps-pone-en-marcha-una-nueva-aplicacion-para-la-comunicacion-de-fabricacion-de-productos-sanitarios-in-house-por-hospitales/
- REGLAMENTO (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) n.o 178/ 2002 y el Reglamento (CE) n.o 1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo (boe.es)
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- UNE-EN ISO 9001:2015. Sistemas de gestión de la calidad. Requisitos (ISO 9001:2015)
- UNE-EN ISO 13485:2018 Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios
- UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN ISO 10993-1:2021 Evaluación biológica de productos sanitarios.
- Directiva 2010/32/UE Protección frente a lesiones por objetos cortopunzantes en el sector sanitario
- Real Decreto 664/1997 modificado por RD 598/2015 Obligación de adoptar medidas técnicas y
- Organizativas específicas para prevenir pinchazos accidentales