4.1 LA COMERCIALIZACIÓN DE PRODUCTOS SANITARIOS EN ESPAÑA: ESTADO ACTUAL Y PERSPECTIVAS
La comercialización de productos sanitarios en España ha experimentado importantes transformaciones en los últimos años, impulsada por los cambios normativos promovidos por la Unión Europea, avances tecnológicos, una creciente demanda en el contexto postpandemia y un entorno de control del gasto público. Para ello, vamos a aproximarnos al estado actual del sector, sus principales desafíos regulatorios, económicos y logísticos, así como las oportunidades de crecimiento e innovación.
Los productos sanitarios abarcan una amplia gama de artículos utilizados en el ámbito médico y hospitalario, desde equipos de diagnóstico hasta dispositivos implantables. En España, su comercialización está sujeta a la normativa europea, en particular al Reglamento (UE) 2017/745 sobre los productos sanitarios (MDR), en vigor desde mayo de 2021. Esta regulación ha supuesto un cambio significativo en los requisitos de evaluación de la conformidad, trazabilidad y vigilancia postcomercialización.
España, como Estado miembro de la Unión Europea, aplica directamente el MDR, lo que implica que todos los productos sanitarios comercializados deben contar con el marcado CE conforme a esta normativa. La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) es el organismo competente en materia de control y supervisión. Desde la entrada en vigor del MDR, se han reforzado los requisitos en cuanto a documentación técnica, análisis clínicos y vigilancia del mercado, con un impacto directo en fabricantes y distribuidores.
Además, el Reglamento (UE) 2017/746 sobre productos sanitarios para diagnóstico in vitro (IVDR), aplicable desde mayo de 2022, ha incrementado también los requisitos específicos para estos dispositivos, afectando a laboratorios y empresas del sector diagnóstico.
Todo ello ha sido desarrollado por el Real Decreto 192/2023 de 21 de marzo, por el que se regulan los productos sanitarios, en los aspectos, que el Reglamento ha dejado a los estados miembros su implementación, atendiendo a sus singularidades. Este RD sustituye el RD 1591/2009, aunque hay aspectos en los que aún sigue en vigor como veremos más adelante.
Asimismo, la Ley 29/2006, de garantías y uso racional de los medicamentos y productos sanitarios, sigue vigente en aspectos complementarios y ha sido modificada parcialmente para adaptarse a las nuevas exigencias europeas.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) es la autoridad competente en materia de control, vigilancia, autorización e inspección de productos sanitarios.
El mercado español de productos sanitarios ha experimentado un crecimiento sostenido en los últimos años. Según datos de la Federación Española de Empresas de Tecnología Sanitaria (FENIN), el sector alcanzó en 2023 una facturación superior a los 9.000 millones de euros. Este crecimiento ha sido impulsado por el envejecimiento poblacional, el incremento de enfermedades crónicas y la modernización tecnológica del sistema de salud.
El ecosistema empresarial está compuesto tanto por filiales de grandes multinacionales como por pymes nacionales. Existe una especialización significativa en productos de un solo uso, implantes ortopédicos, diagnóstico in vitro, y equipos electromédicos. La dependencia de proveedores externos, sin embargo, sigue siendo un reto estructural.
No obstante, el sector enfrenta importantes retos: retrasos en la certificación bajo el MDR debido a la escasez de organismos notificados, dificultades logísticas en la cadena de suministro global y presiones en la financiación pública del sistema sanitario.
El sector está viviendo una transformación significativa gracias a la incorporación de tecnologías emergentes como la inteligencia artificial, el big data, y los dispositivos conectados. Estas innovaciones se han incorporado tanto a dispositivos médicos como a sistemas de monitorización remota y software como producto sanitario (SaMD).
El PERTE de Salud de Vanguardia, aprobado por el Gobierno de España en 2021, contempla inversiones público-privadas por más de 1.500 millones de euros, destinadas a fortalecer la investigación biomédica y la industria de productos sanitarios innovadores.
Uno de los principales retos actuales es el cumplimiento de los requisitos del MDR y el IVDR, especialmente en lo que respecta a la evaluación clínica y la obtención del marcado CE. La escasez de organismos notificados ha provocado cuellos de botella, retrasando la comercialización de numerosos productos.
Los principales retos actuales se centran en la adaptación al entorno regulatorio más estricto, la sostenibilidad de la producción, la dependencia de proveedores externos y la necesidad de fortalecer la industria local. A medio plazo, la implementación completa de la base de datos EUDAMED y la armonización plena de los requisitos facilitarán una mayor transparencia y control.
A nivel operativo, las cadenas de suministro globales continúan mostrando vulnerabilidades, derivadas de la pandemia de COVID-19 y de conflictos geopolíticos que afectan el comercio internacional. Esto ha motivado un renovado interés en la relocalización industrial.
Por otro lado, las oportunidades pasan por la relocalización industrial, la digitalización de procesos, y el fortalecimiento de las sinergias entre sector público y privado.
4.2 LA COMERCIALIZACIÓN DE PRODUCTOS SANITARIOS EN ESPAÑA: NORMATIVA, REQUISITOS Y PAPEL DE LA AEMPS
Se entiende por “comercialización” al suministro de un producto sanitario (PS) o producto sanitario para diagnóstico in vitro, excepto los productos destinados a investigación, para su distribución o uso en el mercado de la Unión. Esto incluye la venta, el alquiler y cualquier otra forma de transferencia, ya sea a título oneroso o gratuito.
La comercialización de productos sanitarios en España se rige por un compendio de normas que tiene como objetivo garantizar la seguridad, eficacia y calidad de éstos. Se regula por el Real Decreto 192/2023, de 21 de marzo y establece el marco normativo nacional para la regulación de los productos sanitarios en España, adaptando la legislación nacional al Reglamento (UE) 2017/745.
Para que un producto sanitario pueda comercializarse en España, es imprescindible que cuente con el marcado CE, que acredita su conformidad con los requisitos esenciales de seguridad y eficacia establecidos por la normativa. Este marcado debe ir acompañado de la declaración de conformidad del fabricante y la documentación técnica pertinente.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) actúa como autoridad competente y tiene un papel clave en la supervisión y control del mercado. Además de velar por la seguridad y eficacia de los productos, la AEMPS establece una serie de requisitos que deben cumplirse para la comercialización de productos sanitarios en España:
- Registro de empresas: Las empresas que fabrican, importan o distribuyen productos sanitarios deben estar inscritas en el Registro de Empresas de Productos Sanitarios (REPS). Este registro garantiza la trazabilidad y control de las empresas que intervienen en la cadena de suministro.
- Comunicación de puesta en el mercado: Antes de comercializar un producto sanitario en España, el fabricante o su representante autorizado debe notificar a la AEMPS la puesta en el mercado del producto. Esta notificación se realiza a través del Registro de Productos Sanitarios (RPS), que recopila información clave sobre los productos disponibles.
- Sistema de vigilancia y notificación: Las empresas están obligadas a implementar un sistema de vigilancia poscomercialización y a notificar a la AEMPS cualquier incidente adverso o defecto de calidad detectado. Esto permite identificar riesgos potenciales y adoptar medidas correctivas si es necesario.
- Etiquetado e instrucciones de uso: La AEMPS exige que los productos sanitarios cuenten con un etiquetado y unas instrucciones de uso claros, precisos y en castellano. Esta información debe proporcionar las advertencias necesarias y detallar la forma de utilización segura del producto.
- Cumplimiento de la publicidad: La publicidad de los productos sanitarios debe respetar los principios de veracidad y no inducir a error. La AEMPS supervisa que la información publicitaria no incluya afirmaciones engañosas ni exagere las propiedades del producto.
Registros actuales en vigor en la AEMPS
- Comunicación de Comercialización de Productos Sanitarios (CCPS): en el que los fabricantes, representantes autorizados o importadores deben notificar la puesta en el mercado de sus productos sanitarios en España. Tiene como objetivo permitir a la AEMPS y a las comunidades autónomas supervisar y garantizar la seguridad y eficacia de los productos comercializados. Es un requisito obligatorio antes de comercializar el producto sanitario, pero no implica una autorización previa de comercialización, sino una notificación.
- Responsables de productos sanitarios: Es un registro oficial de la AEMPS en el que se inscriben las empresas y entidades que fabrican, importan o distribuyen productos sanitarios en España. Asegura la trazabilidad y control de los productos y de las actividades realizadas por los agentes económicos. Es obligatorio para operar legalmente en el sector y ayuda a garantizar la seguridad y calidad de los productos comercializados.
4.3 CÓMO REGULA EL REGLAMENTO (UE) 2017/745 LA COMERCIALIZACIÓN DE PRODUCTOS SANITARIOS EN LA UNIÓN EUROPEA
El Reglamento 2017/745 establece un marco legal armonizado para la comercialización, puesta en servicio y uso de productos sanitarios en la UE, aplicable a todos los Estados miembros.
Requisitos para comercializar productos sanitarios
- Marcado CE: El producto debe haber pasado por el procedimiento de evaluación de la conformidad correspondiente a su clase de riesgo (I, IIa, IIb o III). Tras superar este procedimiento, el fabricante colocará el marcado CE, que acredita que el producto cumple con los requisitos esenciales de seguridad y rendimiento.
- Documentación técnica y declaración de conformidad: El fabricante debe elaborar y conservar la documentación técnica del producto. Debe emitir una declaración UE de conformidad, que formaliza su compromiso de que el producto cumple con el MDR.
- Sistemas de vigilancia y trazabilidad: Los agentes económicos (fabricantes, importadores y distribuidores) están obligados a implementar sistemas para:
- Vigilancia poscomercialización (recoger y evaluar información sobre la seguridad y el rendimiento del producto una vez comercializado).
- Trazabilidad (uso del sistema UDI para rastrear los productos desde el fabricante hasta el usuario final).
Obligaciones de los agentes económicos
El MDR establece obligaciones específicas para cada agente económico:
- Fabricantes: Diseñar, fabricar y comercializar productos que cumplan con los requisitos generales de seguridad y rendimiento. Garantizar la vigilancia y el control de los productos tras su comercialización.
- Representantes autorizados (cuando el fabricante no está en la UE): Actuar como punto de contacto con las autoridades competentes. Velar por la conformidad de los productos importados.
- Importadores: Comprobar que los productos cuentan con el marcado CE y la declaración de conformidad. Garantizar que la información del etiquetado e instrucciones esté en el idioma oficial del país.
- Distribuidores: Comprobar que los productos cuentan con la documentación requerida. Garantizar que las condiciones de almacenamiento y transporte no afecten la conformidad.
Puesta en el mercado y puesta en servicio
El MDR distingue entre:
- Puesta en el mercado: Momento en que el producto se suministra para su distribución o uso por primera vez en la UE.
- Puesta en servicio: Momento en que el producto está disponible para su uso final en la UE.
Ambos conceptos están sujetos a las normas y obligaciones del MDR.
Prohibiciones y restricciones
El Reglamento establece la prohibición de comercializar productos no conformes o que representen un riesgo para la salud o la seguridad de los usuarios.
Además, otorga a los Estados miembros la potestad de:
- Adoptar medidas para restringir o prohibir la comercialización de productos por razones de salud pública.
- Realizar inspecciones y controles de mercado para garantizar el cumplimiento.
Evaluación de la conformidad y control de mercado
- Organismos notificados: Certifican la conformidad de los productos de mayor riesgo (clases IIa, IIb y III).
- Autoridades competentes: Supervisan que todos los agentes económicos cumplen sus obligaciones.
4.4 CÓMO REGULA EL REAL DECRETO 192/2023 LA COMERCIALIZACIÓN DE PRODUCTOS SANITARIOS EN EL TERRITORIO ESPAÑOL
El Capítulo V del Real Decreto 192/2023, de 21 de marzo, establece las disposiciones relativas a la comercialización y puesta en servicio de productos sanitarios en España. Este capítulo complementa el Reglamento (UE) 2017/745, adaptando ciertos aspectos al contexto nacional.
- Registro de comercialización (Artículo 18): Se crea un Registro de Comercialización gestionado por la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). En él deben inscribirse los agentes económicos que comercialicen productos sanitarios (excepto productos a medida) en España. Esta inscripción es obligatoria antes de iniciar la actividad comercial.
- Identificación y trazabilidad (Artículo 19): Los productos deben cumplir con los requisitos de identificación y trazabilidad, incluyendo el sistema UDI (Identificación Única de Producto), conforme al Reglamento (UE) 2017/745. Esto garantiza la trazabilidad desde el fabricante hasta el usuario final.
- Reetiquetado, reenvasado y traducciones (Artículo 20): Las operaciones de reetiquetado, reenvasado o traducción de la documentación deben ser notificadas a la AEMPS. Estas actividades deben realizarse sin alterar las condiciones originales del producto y asegurando que la información proporcionada sea precisa y actualizada.
- Registro de responsables de productos a medida (Artículo 21): Se establece un Registro de Responsables de la puesta en el mercado de productos a medida, donde deben inscribirse los fabricantes de estos productos. Este registro permite a la AEMPS supervisar la fabricación y comercialización de productos personalizados.
- Información a las comunidades autónomas (Artículo 22): La AEMPS debe informar a las comunidades autónomas sobre los productos comercializados en su territorio, facilitando la coordinación y vigilancia del mercado en las comunidades autónomas.
- Obligaciones de los agentes económicos (Artículo 23): Los fabricantes, importadores y distribuidores deben cumplir con las obligaciones establecidas en el Reglamento (UE) 2017/745, incluyendo la garantía de conformidad de los productos, la gestión de riesgos y la cooperación con las autoridades competentes.
- Distribución y venta (Artículos 24 y 25): Se regulan las condiciones para la distribución y venta de productos sanitarios, exigiendo que los distribuidores cuenten con sistemas de calidad adecuados y que los productos se almacenen y transporten en condiciones que no comprometan su calidad ni seguridad.
- Establecimientos de venta al público (Artículo 26): Los establecimientos que venden productos que requieren adaptación individualizada deben contar con la autorización correspondiente y cumplir con los requisitos específicos establecidos por la normativa.
- Exhibiciones (Artículo 27): Se permite la exhibición de productos sanitarios no conformes (por ejemplo, sin marcado CE) en ferias y exposiciones, siempre que se indique claramente que no están disponibles para la venta ni para su uso clínico. Este capítulo refuerza la vigilancia y control del mercado de productos sanitarios en España, asegurando que los productos comercializados cumplan con los estándares de seguridad y eficacia establecidos por la normativa europea y nacional.
El Real Decreto 192/2023, de 21 de marzo, regula los productos sanitarios en España, adaptando la normativa nacional al Reglamento (UE) 2017/745. Aunque deroga en gran medida el Real Decreto 1591/2009, de 16 de octubre, algunas de sus disposiciones permanecen vigentes de forma transitoria hasta que se desarrollen normativas específicas.
Aspectos pendientes de regulación
- Publicidad y promoción de productos sanitarios: Los artículos 38 a 40 del Real Decreto 1591/2009, que regulan la publicidad, promoción, incentivos y patrocinio de reuniones científicas, siguen en vigor hasta que se apruebe una normativa específica que los sustituya.
- Regulación de productos sanitarios para diagnóstico in vitro: El Real Decreto 192/2023 no aborda específicamente estos productos, que están regulados por el Reglamento (UE) 2017/746. Se espera una normativa nacional que complemente este reglamento europeo.
- Procedimientos técnicos para el reprocesamiento de productos de un solo uso: Aunque el Real Decreto 192/2023 introduce la posibilidad de reprocesar estos productos, los procedimientos y requisitos específicos para llevar a cabo esta actividad aún deben ser desarrollados por el Ministerio de Sanidad.
- Adaptación del Real Decreto Legislativo 1/2015: Este texto refundido de la Ley de garantías y uso racional de los medicamentos y productos sanitarios aún no ha sido adaptado completamente para alinearse con las disposiciones del Real Decreto 192/2023 y los reglamentos europeos.
- Evaluación de tecnologías sanitarias: Aunque se ha avanzado en la elaboración de un proyecto de Real Decreto sobre Evaluación de Tecnologías Sanitarias (ETS), este aún no ha sido aprobado. Su implementación es clave para la adopción de innovaciones y la mejora del sistema sanitario.
4.5 PUESTA EN EL MERCADO ESPAÑOL DE UN PRODUCTO – REGULACÍON Y REQUISITOS
La puesta en el mercado de productos sanitarios en España se enmarca dentro del contexto normativo de la Unión Europea, establecido principalmente por el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, relativo a los productos sanitarios (en adelante, MDR), el cual es de aplicación directa en todos los Estados miembros. Este reglamento ha sido complementado a nivel nacional por el Real Decreto 192/2023, que adapta e implementa los requisitos europeos en el ordenamiento jurídico español. El cumplimiento de este marco normativo es imprescindible para garantizar la seguridad, calidad y eficacia de los productos sanitarios antes de su comercialización en el mercado español.
1. Clasificación del producto sanitario
El primer paso en el proceso de puesta en el mercado consiste en determinar la clase de riesgo del producto, conforme a las reglas establecidas en el Anexo VIII del MDR. Esta clasificación (clase I, IIa, IIb o III) está basada en el grado de invasividad del producto, su duración de uso y su función terapéutica o diagnóstica. La clasificación influye de manera determinante en la vía de evaluación de la conformidad y en los requisitos de documentación técnica exigidos.
2. Evaluación de la conformidad
Una vez determinada la clase del producto, el fabricante debe someterse a un procedimiento de evaluación de la conformidad, con el objetivo de demostrar que el producto cumple con los requisitos esenciales del MDR. Para los productos de clase I (no estériles y sin función de medición), el fabricante puede realizar una autoevaluación. Sin embargo, para las demás clases (I estéril, I con función de medición, IIa, IIb y III), es obligatorio acudir a un organismo notificado (ON) que supervise y valide el proceso.
3. Evaluación clínica
Como parte del proceso de conformidad, el fabricante debe realizar una evaluación clínica del producto, en la que se analicen datos clínicos procedentes de la literatura científica, de productos equivalentes ya comercializados o de ensayos clínicos propios. Esta evaluación debe ser proporcional al riesgo del producto y se integra en el expediente técnico que respalda el marcado CE. Todo ello viene especificado en los anexos I, II, III y XIV del MDR.
- Anexo I: Requisitos generales de seguridad y funcionamiento
- Anexo II: Documentación técnica
- Anexo III: Documentación técnica sobre seguimiento poscomercialización
- Anexo XIV: Evaluación clínica y seguimiento clínico poscomercialización
4. Marcado CE
Superado el procedimiento de evaluación de la conformidad, el producto podrá llevar el marcado CE, el cual indica que cumple con todos los requisitos reglamentarios aplicables. El marcado debe ser visible, legible e indeleble y, en caso de que haya intervenido un organismo notificado, debe incluir su número de identificación.
5. Registro y comunicación a las autoridades competentes
En el caso español, antes de la puesta en el mercado de productos de clase IIa, IIb y III, el fabricante (o su representante autorizado, si se encuentra fuera de la UE) debe realizar una comunicación previa a la Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) a través del portal correspondiente. Además, los agentes económicos (fabricantes, importadores, distribuidores y representantes autorizados) deben inscribirse en el Registro de responsables de la comercialización de productos sanitarios.
De forma complementaria, tanto el producto como los agentes económicos deben registrarse en la base de datos europea EUDAMED, una vez que esta esté completamente operativa.
6. Representante autorizado en la UE
Cuando el fabricante no está establecido en un Estado miembro de la Unión Europea, es obligatorio designar un representante autorizado en la UE que actúe como interlocutor ante las autoridades nacionales y garantice el cumplimiento de las obligaciones derivadas del MDR.
7. Sistema de gestión de calidad
Especialmente para productos de clases superiores (IIa, IIb y III), es requisito contar con un sistema de gestión de calidad, habitualmente conforme con la norma internacional UNE-EN ISO 13485, que asegure la trazabilidad, el control de la producción y la vigilancia postcomercialización.
8. Vigilancia postcomercialización
El fabricante debe implantar un sistema de vigilancia postcomercialización (PMS) que permita recopilar, evaluar y, en su caso, actuar frente a cualquier incidente o desviación detectada durante el uso del producto. La notificación de incidentes graves o medidas correctivas de seguridad a la AEMPS es obligatoria, conforme a lo previsto en el MDR y en el Real Decreto 192/2023.
9. Etiquetado e instrucciones de uso
Finalmente, todos los productos sanitarios deben ir acompañados de un etiquetado y un manual de instrucciones de uso redactados al menos en castellano, que incluyan información clara y comprensible sobre el uso previsto, precauciones, contraindicaciones, condiciones de almacenamiento, trazabilidad y otros aspectos esenciales.
CONCLUSIONES
La comercialización de productos sanitarios en España se encuentra en un momento de transformación. Si bien el nuevo marco regulatorio plantea desafíos significativos, también abre la puerta a un sector más seguro, competitivo e innovador. La apuesta por la fabricación nacional, la digitalización y la cooperación internacional serán claves para asegurar la sostenibilidad y la autonomía estratégica del sistema sanitario español. El cumplimiento normativo, la digitalización y la colaboración entre administraciones y empresas serán factores clave para un desarrollo sostenible y competitivo del sector.
4.5 VIGILANCIA POST-COMERCIALIZACIÓN
La vigilancia post-comercialización (Post-Market Surveillance, PMS) constituye un componente esencial del ciclo de vida de los productos sanitarios. Su razón de ser se fundamenta en la gestión de riesgos inherente a los sistemas complejos, en la necesidad de mantener actualizado el perfil beneficio-riesgo de los productos en condiciones reales de uso y en la aplicación de principios de salud pública para la protección de pacientes y usuarios.
Los productos sanitarios no deberían presentar incidencias ya que tienen muchos filtros para comprobar su calidad, pero en el caso de detectarlas se debe actuar de forma urgente para evitar problemas en la salud de los pacientes o profesionales. El entorno sanitario actual con la rápida evolución tecnológica de los dispositivos médicos y por su creciente complejidad técnica plantea nuevos desafíos para los sistemas de vigilancia post-comercialización, que deben ser capaces de:
- Gestionar grandes volúmenes de información procedente de múltiples fuentes.
- Detectar patrones de riesgo emergentes en tiempo oportuno.
- Coordinarse eficazmente entre las distintas partes implicadas: fabricantes, autoridades sanitarias, organismos notificados, centros hospitalarios y profesionales de la salud.
- Incorporar tecnologías de análisis predictivo y vigilancia activa para anticiparse a potenciales problemas de seguridad.
- Ejecutar acciones correctivas, modificación de producto o restricción de uso
- Actualizando procedimientos, estándares de seguridad y regulaciones.
En este sentido, la vigilancia post-comercialización no sólo actúa como mecanismo de control correctivo, sino también como instrumento estratégico de gestión proactiva de riesgos sanitarios siendo un proceso dinámico y adaptativo, esencial en entornos sanitarios complejos y de alta incertidumbre.
Los incidentes adversos por productos sanitarios, pueden tener consecuencias graves para la seguridad de los pacientes y la salud pública. Ejemplos conocidos son los problemas de las prótesis mamarias PIP o las complicaciones de ciertos desfibriladores implantables defectuosos, esto pone de manifiesto la necesidad de un sistema de vigilancia post-comercialización fuerte. Por ello como decíamos en el capítulo de ciclo de la vida de un producto, el control de los productos sanitarios no concluye con su autorización para la comercialización, sino que se prolonga a lo largo de todo su ciclo de vida. Tras su introducción en el mercado, los productos sanitarios se enfrentan a condiciones reales de uso que pueden revelar problemas de seguridad y funcionamiento no detectados durante las fases precomerciales de evaluación clínica y técnica. Por este motivo, la vigilancia post-comercialización es una herramienta esencial para garantizar la seguridad, eficacia y desempeño continuo en condiciones reales. Esto queda recogido en el capítulo VII artículos 83 al 100 y anexos III y XIV del Reglamento 2017/745 (MDR) para proteger a los pacientes y asegurar la confianza en el sistema sanitario, promoviendo una mejora continua en la calidad y seguridad de los productos sanitarios.
El proceso incluye la recopilación de datos sobre el rendimiento del dispositivo a lo largo de su vida útil, utilizando herramientas como informes de incidentes, estudios de seguimiento clínico postcomercialización (PMCF) y retroalimentación de usuarios. Esto permite detectar fallos no previstos durante las fases de diseño y validación, así como evaluar si los beneficios continúan superando los riesgos.
Los objetivos esenciales de la vigilancia postcomercialización son:
- detectar problemas de funcionamiento o seguridad no identificados en su fase de desarrollo.
- permitir realizar la notificación y análisis de incidentes.
- poder realizar la reevaluación del perfil riesgo beneficio.
- informar a las autoridades sanitarias, profesionales y usuarios para mantener la seguridad de los productos.
- promover acciones correctivas o preventivas para minimizar riesgos actualizando la información sobre el diseño, fabricación, instrucciones de uso y etiquetado.
Las obligaciones de los operadores económicos según el Reglamento son:
- Los fabricantes están obligados a implementar un sistema proactivo de vigilancia que documente los eventos adversos graves y establezca medidas correctivas, como modificaciones en el diseño o actualizaciones en las instrucciones de uso según la clase de riesgo. Además, de los productos de clase IIa, IIb y III deben actualizar periódicamente el informe de seguridad postcomercialización (PSUR) los de clase IIa como mínimo cada dos años y los de clase IIb y III cada año salvo en los productos a medida que el PSUR formara parte de la documentación técnica. El PSUR proporciona un análisis detallado del rendimiento del producto en el mercado.
- Los distribuidores tienen la obligación de informar a los fabricantes y autoridades sanitarias de cualquier incidente conocido.
- Los importadores asumen responsabilidades similares a las de los distribuidores y deben verificar que los productos cumplen con la normativa vigente antes de su comercialización.
El Reglamento (UE) 2017/746 de los productos sanitarios para diagnóstico in vitro y establece disposiciones equivalentes en materia de vigilancia post-comercialización. Este reglamento impone requisitos específicos para la recogida, análisis y notificación de incidentes adversos relacionados con dispositivos de laboratorio y test diagnósticos, garantizando así su seguridad continuada.
La base de datos EUDAMED está diseñada para ser interoperable con los sistemas de vigilancia de terceros países, diferentes de la UE y organizaciones internacionales, favoreciendo una supervisión global coordinada.
4.6 SEGUIMIENTO Y REPORTE DE INCIDENTES. AGENCIA ESPAÑOLA DE MEDICAMENTOS Y PRODUCTOS SANITARIOS. ALERTAS SANITARIAS
Según se recoge en el Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo, de 5 de abril de 2017, Sección 2 Vigilancia, articulo 87 Notificación de incidentes graves y acciones correctivas de seguridad y en el RD 192/2023 de 21 de marzo, por el que se regulan los productos sanitarios, en el artículo 35 Sistema de vigilancia tanto los fabricantes como el personal sanitario y pacientes pueden realizar una notificación. Los fabricantes deberán realizar la notificación en el sistema electrónico creado para tal fin por la Comisión europea mientras que los profesionales y pacientes lo realizarán en la Agencia Española de Medicamentos y Productos Sanitarios que lo trasladara al fabricante del producto afectado.
Los fabricantes informarán de cualquier incidente grave, excepto los efectos secundarios que ya estén documentados en la información del producto que deben incluirse en la notificación de tendencias, inmediatamente después de que haya establecido la relación de causalidad entre dicho incidente y su producto o haya establecido que dicha relación de causalidad es razonablemente posible, y a más tardar quince días después de tener conocimiento del incidente. En el caso de una amenaza grave para la salud pública se deberá realizar en el plazo de dos días. Cuando sea preciso para garantizar la notificación rápida, el fabricante podrá presentar un informe preliminar incompleto, seguido de un informe completo.
Los fabricantes informarán a la AEMPS de cualquier acción correctiva de seguridad antes de que tal acción se lleve a cabo. Asimismo, remitirán la nota de seguridad prevista para su comunicación a los usuarios o clientes antes de su difusión. Esta nota de seguridad deberá facilitarse en castellano. La AEMPS podrá determinar la conveniencia de ejecutar las medidas propuestas, impidiéndolas o modificándolas por razones justificadas de salud pública.
El profesional que detecte la incidencia la debe notificar a los encargados de vigilancia para los procedimientos de notificación de los incidentes del producto sanitario del centro. En unos centros suele ser algún representante de la dirección médica, jefe de farmacia, supervisor de RRMM o jefe de compras. La notificación debe ser siempre por escrito, de la forma más exhaustiva posible ya que los responsables deben valorar realizar una comunicación escrita a la empresa suministradora para poder solventar el problema y dependiendo de la implicación notificar a la AEMPS, para que ellos después de un seguimiento decidan si el problema es susceptible de realizar una alerta sanitaria.
Los centros sanitarios deben designar un responsable de vigilancia para los productos sanitarios que tenga trato directo con la AEMPS. Comunicará sus datos a las autoridades sanitarias de la correspondiente Comunidad Autónoma y a la AEMPS. Este responsable puede ser el mismo o no, que el encargado interno. Las notificaciones emitidas por la AEMPS les llegarán a ellos siempre. Las notificaciones deben realizarse en el punto de contacto de la Comunidad Autónoma correspondiente, siguiendo las directrices para la aplicación del sistema de vigilancia por los centros y profesionales sanitarios.
El informe deberá incluir toda la información del producto: nombre, referencia, número de lote, fecha de fabricación, fecha de caducidad, y explicación precisa del problema detectado, indicándose si se ha producido en la apertura o en su uso. Si el envoltorio no lo hemos desechado previamente también se debe remitir con el informe de la incidencia.
Para realizar la notificación la AEMPS tiene habilitada a través de su sede electrónica un formulario. Partiendo de la pagina principal existe un espacio llamado notifica para los diferentes medicamentos y productos:
- Notifica RAM para medicamentos
- Notifica VET para medicamentos veterinarios
- Notifica PS para productos sanitarios
- Notifica CS para productos cosméticos.

Pinchando en Notifica PS llegas a esta pantalla
Desde este punto, guía a otra pantalla para rellenar la notificación solicitándote si es profesional sanitario y Comunidad Autónoma.
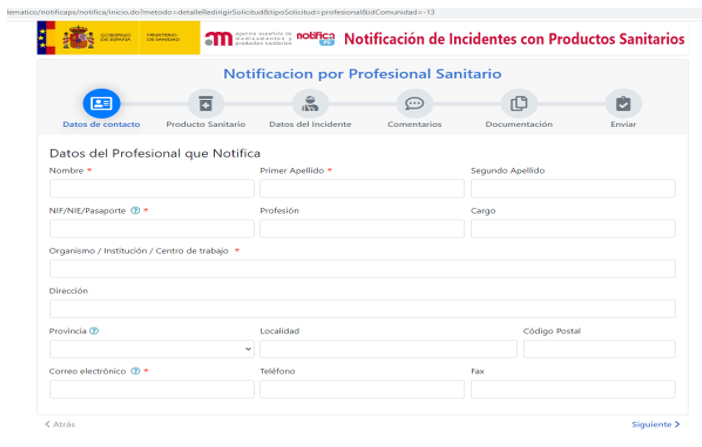
Posteriormente te llevar a una página donde se debe rellenar los datos del producto sanitario afectado.
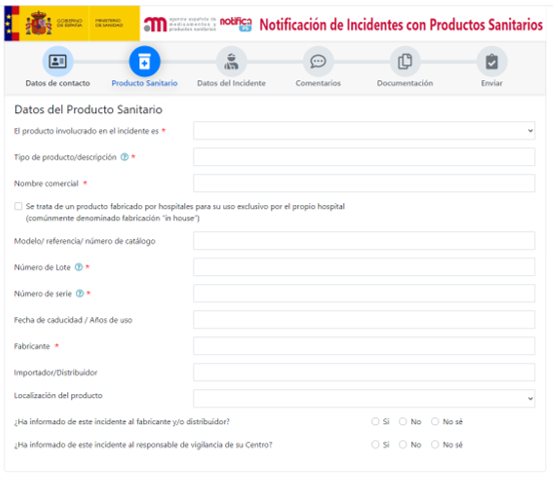
También se deberá rellanar datos del incidente, comentarios y da la posibilidad de incluir documentos adjuntos.
Los pacientes y usuarios también podrán notificar los incidentes graves a la AEMPS utilizando el procedimiento electrónico habilitado para ello, sin perjuicio de la notificación que hayan podido hacer al fabricante, o a otro agente económico, o al profesional sanitario. La AEMPS coordinará con las comunidades autónomas, a través del mencionado procedimiento electrónico, la recepción de las notificaciones recibidas por los profesionales sanitarios, los pacientes o los usuarios.
La AEMPS evaluará y adoptara las medidas necesarias de protección de la salud y dará conocimiento a las autoridades sanitarias de las comunidades autónomas y a la Inspección General de Sanidad de la Defensa de la información relativa a las medidas adoptadas, o que corresponda adoptar, en relación con las notificaciones de vigilancia. Igualmente dará conocimiento a otros agentes afectados en los casos en que resulte oportuno.
Las incidencias pueden estar debidas por problemas de funcionamiento, alteración de las características o de las prestaciones, deficiencias en el etiquetado, en las instrucciones de utilización de los productos o incumplimiento respecto a la legislación, que pueden suponer un riesgo para la salud de los pacientes tratados con los mismos o para los profesionales. Las empresas también deberán comunicar las medidas correctoras que procedan.
La AEMPS registrará de forma centralizada las notificaciones recibidas, tanto por los responsables como cualquier otro personal siempre que estén debidamente justificadas. A través de Unidad de Vigilancia de Productos Sanitarios se evaluará el problema y generará la Alerta de Productos Sanitarios, adoptando en caso necesario las medidas necesarias de protección de la salud. Estas medidas se transmitirán a los puntos de contacto de vigilancia de productos sanitarios de las Comunidades Autónomas y a la Inspección General de Sanidad de la Defensa para su difusión a los centros de su ámbito territorial. También informará a la Comisión Europea y al resto de Estados miembros de las medidas adoptadas o previstas para minimizar la repetición de los incidentes notificados, incluyendo la información relativa a tales incidentes. Siempre que sea posible se realizará la comunicación conjuntamente con el fabricante o su representante autorizado.
La difusión en las comunidades se suele realizar de forma de cascada, de manera que la dirección de sanidad comunica a los gerentes de los centros, estos a los responsables de vigilancia. La urgencia y la amplitud de su difusión se realiza teniendo en cuenta la gravedad o riesgo que supone el problema detectado, la probabilidad de que se presente de nuevo, la distribución del producto sanitario, la información facilitada por el fabricante o distribuidor y las medidas que ha adoptado. etc.
BIBLIOGRAFÍA
- https://es.linkedin.com/pulse/el-instrumental-quir%C3%BArgico-algo-de-su-historia-albarracin-miranda.
- historia.nationalgeographic.com.es/a/sofisticacion-antiguo-egipto-protesis-hace-3000-anos_11639
- portalcomunicacion.uah.es/diario-digital/actualidad/medicina-medieval-una-etapa-oscura-pero-llena-de-supersticiones/
- www.salusplay.com/apuntes/quirofano-y-anestesia/tema-3-instrumentacion
- wfsahq.org/es/about/history/history-of-anaesthesia
- www.bbvaopenmind.com/ciencia/investigacion/joseph-lister-el-hombre-que-esterilizo-la-cirugia/
- www.revistamedicinaycultura.fmposgrado.unam.mx/index.php/2023/01/24/joseph-lister-biografia/
- identyd.com/historia-autoclave/
- www.elsevier.es/es-revista-revista-argentina-radiologia-383-articulo-wilhelm-conrad-roentgen-el-descubrimiento-S0048761916301545
- www.fda.gov/medical-devices/overview-device-regulation/history-medical-device-regulation-oversight-united-states
- formasefh.sefh.es/tecnifarmh/curso-productos-sanitarios/curso-productos-sanitarios.pdf
- www.aemps.gob.es/la-aemps/legislacion/legislacion-sobre-productos-sanitarios/
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- www.aemps.gob.es/productos-sanitarios/investigacionclinica-productossanitarios/
- UNE-EN ISO 14971:2020 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN 62366-1:2015/A1:2020 (Ratificada) Productos sanitarios. Parte 1: Aplicación de la ingeniería de usabilidad a los productos sanitarios.
- Aplicación de la gestión de riesgos a los productos sanitarios (une.org)
- 231219_GuiaValidacion_Tec_Sanitarias-1.pdf (inndromeda.es)
- Ciclo de vida de producto sanitario archivos - Consultoría de producto sanitario
- Ciclo de vida de producto sanitario y gestión de riesgos - Fernando Atienza
- Producto sanitario definición, características y usos (productossanitarios.net)
- Risk Management in the Medical Device Industry (dqsglobal.com)
- Design Thinking en Producto Sanitario ¿Qué es idear, prototipar y testar? (ambit-bst.com)
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Ley 29/2006, de 26 de julio de garantías y uso racional del medicamento.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS): https://www.aemps.gob.es/
- Federación Española de Empresas de Tecnología Sanitaria (FENIN): https://www.fenin.es/
- Ministerio de Ciencia e Innovación (PERTE Salud de Vanguardia): https://www.ciencia.gob.es/
- European Commission. (2021). Guidance on the application of the MDR and IVDR during the COVID-19 pandemic. Publications Office of the European Union. https://health.ec.europa.eu/system/files/2021-05/md_guidance_md_011_en_0.pdf
- Ghisellini, P., Cialani, C., & Ulgiati, S. (2016). A review on circular economy: The expected transition to a balanced interplay of environmental and economic systems. Journal of Cleaner Production, 114, 11-32. https://doi.org/10.1016/j.jclepro.2015.09.007
- Organización Internacional del Trabajo (OIT). (2023). Declaración de principios y derechos fundamentales en el trabajo. https://www.ilo.org/global/lang--es/index.htm
- Organización Mundial de la Salud (OMS). (2022). Health product policy and standards. https://www.who.int/teams/health-product-and-policy-standards
- Parlamento Europeo y Consejo de la Unión Europea. (2017). Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo de 5 de abril de 2017 sobre los productos sanitarios. Diario Oficial de la Unión Europea. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX%3A32017R0745
- Porter, M. E., & Kramer, M. R. (2011). Creating shared value. Harvard Business Review, 89(1/2), 62-77.
- OMS. (2015). Ética de la salud mundial: cuestiones clave.
- Declaración de Helsinki de la AMM – Principios éticos para las investigaciones médicas con participantes humanos – WMA – The World Medical Association.
- Global health ethics: key issues (who.int)
- https://lac.saludsindanio.org/cambio-climatico-y-salud/huella-climatica-del-sector-salud
- https://www.miteco.gob.es/content/dam/miteco/es/cambio-climatico/temas/mitigacion-politicas-y-medidas/docuementoexplicativosello_tcm30-479000.pdf
- https://accionclimaticaensalud.org/sites/default/files/2021-06/huellaclimatica.pdf
- Estrategia de Economía Circular de Euskadi 2030 - Prevención de la contaminación, inspección y control ambiental - Euskadi.eus
- Residuos Sanitarios (miteco.gob.es)
- Guía de procedimientos de esterilización en el medio hospitalario. file:///C:/Users/50820026P/Documents/OneDrive%20-%20Madrid%20Digital/PERSONAL/MASTER%20ordenador%20sollube/2.%20TEMARIO%20MASTER%20FARMA%20Y%20PS/procedementos_esterilizacion.pdf
- Curso básico de Gestion de Productos Sanitarios ANECORM
- https://www.aemps.gob.es/informa/la-aemps-pone-en-marcha-una-nueva-aplicacion-para-la-comunicacion-de-fabricacion-de-productos-sanitarios-in-house-por-hospitales/
- REGLAMENTO (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) n.o 178/ 2002 y el Reglamento (CE) n.o 1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo (boe.es)
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- UNE-EN ISO 9001:2015. Sistemas de gestión de la calidad. Requisitos (ISO 9001:2015)
- UNE-EN ISO 13485:2018 Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios
- UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN ISO 10993-1:2021 Evaluación biológica de productos sanitarios.
- Directiva 2010/32/UE Protección frente a lesiones por objetos cortopunzantes en el sector sanitario
- Real Decreto 664/1997 modificado por RD 598/2015 Obligación de adoptar medidas técnicas y
- Organizativas específicas para prevenir pinchazos accidentales