5.1 NORMATIVA Y RECOMENDACIONES INTERNACIONALES SOBRE LA CALIDAD EN LOS PRODUCTOS SANITARIOS
La calidad en los productos sanitarios constituye un pilar esencial para garantizar la seguridad, eficacia y desempeño de estos dispositivos dentro del sistema de salud. Debido a la creciente complejidad tecnológica de los productos sanitarios y su trascendencia en la atención médica, los organismos nacionales e internacionales han desarrollado marcos regulatorios y normativos robustos con el objetivo de proteger a los pacientes y asegurar altos estándares de fabricación, control y uso clínico. A continuación. se examinan los principales marcos normativos internacionales y las recomendaciones que orientan la gestión de la calidad en productos sanitarios, así como su implementación en los sistemas regulatorios nacionales.
5.1.1 Concepto de calidad en los productos sanitarios
La calidad en los productos sanitarios se refiere a su capacidad para cumplir con los requisitos establecidos —legales, normativos y clínicos— durante todo su ciclo de vida, desde el diseño y desarrollo, hasta su fabricación, comercialización, uso y retirada del mercado. La calidad está intrínsecamente relacionada con la seguridad del paciente, el cumplimiento de la función prevista y la reducción de riesgos asociados a su uso.
La Organización Mundial de la Salud (OMS) y organismos como la Organización Internacional de Normalización (ISO) han promovido definiciones comunes y armonizadas que vinculan la calidad con principios de gestión del riesgo, mejora continua y vigilancia poscomercialización.
5.1.2 Principales marcos normativos internacionales
Reglamento (UE) 2017/745 sobre los productos sanitarios (MDR)
En la Unión Europea, el Reglamento (UE) 2017/745 reemplazó a la Directiva 93/42/CEE con el objetivo de fortalecer el sistema regulador y aumentar la transparencia, trazabilidad y vigilancia de los productos sanitarios. Este reglamento establece exigencias detalladas en cuanto a:
- Clasificación y evaluación de la conformidad.
- Documentación técnica y expediente del producto.
- Gestión de riesgos y validación clínica.
- Sistema de calidad del fabricante.
- Requisitos para los organismos notificados.
- Sistema de vigilancia postcomercialización y seguimiento clínico posterior a la comercialización (PMCF).
El MDR también pone un fuerte énfasis en la responsabilidad del fabricante y la trazabilidad de los productos mediante el sistema de identificación única (UDI).
Normativa estadounidense (FDA – 21 CFR Part 820)
En Estados Unidos, la Administración de Alimentos y Medicamentos (FDA) regula los productos sanitarios a través del título 21 del Código de Regulaciones Federales (21 CFR). La parte 820 establece los requisitos del sistema de calidad (QSR), que incluyen:
- Procedimientos de control de diseño.
- Validación de procesos.
- Control de cambios.
- Registros de calidad y producción.
- Gestión de no conformidades y acciones correctivas y preventivas (CAPA).
La FDA ha anunciado una armonización progresiva con la norma ISO 13485, reflejando una tendencia hacia la convergencia internacional.
Norma UNE-EN ISO 13485:2018 Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios. (ISO 13485:2016). (Versión consolidada)
La norma ISO 13485 es el estándar internacional más ampliamente reconocido para los sistemas de gestión de calidad aplicables a productos sanitarios. Define requisitos específicos que incluyen:
- Control documental y de registros.
- Responsabilidad de la dirección y gestión de recursos.
- Diseño y desarrollo.
- Producción y provisión del servicio.
- Vigilancia, acciones correctivas y mejora continua.
A diferencia de ISO 9001, ISO 13485 enfatiza la gestión del riesgo, la validación de procesos y la trazabilidad, elementos críticos en el sector de productos sanitarios.
Esquema IMDRF y su influencia en la regulación global
El Foro Internacional de Reguladores de Productos Sanitarios (IMDRF), sucesor del GHTF (Global Harmonization Task Force), promueve la armonización de marcos regulatorios mediante documentos guía. Entre sus contribuciones más relevantes se encuentran:
- Definición de criterios para la clasificación de productos sanitarios.
- Guías sobre el ciclo de vida del software como producto sanitario.
- Buenas prácticas de evaluación clínica.
- Recomendaciones sobre auditorías y vigilancia postmercado.
Los documentos de IMDRF sirven como referencia para muchas autoridades regulatorias, incluyendo países de Latinoamérica, Asia y África que están en proceso de desarrollar o fortalecer sus marcos normativos.
5.1.3 Recomendaciones de la OMS y organismos multilaterales: Rol de la OMS en el control de calidad de productos sanitarios
La Organización Mundial de la Salud (OMS) desempeña un papel fundamental en el fortalecimiento de los sistemas de salud a nivel global, y dentro de ello, en la mejora del acceso, la seguridad y la calidad de los productos sanitarios. Aunque no actúa como autoridad regulatoria vinculante, la OMS proporciona marcos técnicos, recomendaciones estratégicas, herramientas normativas y mecanismos de evaluación que sirven de referencia especialmente para países con sistemas regulatorios en desarrollo o en proceso de armonización.
Rol de la OMS en productos sanitarios: visión general
La OMS define los productos sanitarios como cualquier instrumento, aparato, implemento, máquina o software destinado a ser utilizado con fines médicos en el diagnóstico, tratamiento, monitoreo o prevención de enfermedades. En este contexto, la OMS actúa en tres niveles:
- Normativo y técnico: establece estándares, guías y marcos de referencia.
- Operativo: apoya a los Estados Miembros en la implementación de políticas nacionales.
- Evaluación y precalificación: coordina mecanismos para garantizar la calidad de productos esenciales.
Publicaciones técnicas y guías normativas: Documento marco sobre productos sanitarios
La OMS ha publicado documentos clave que orientan a los países en la regulación, adquisición y uso de productos sanitarios. Entre ellos:
- "WHO Global Model Regulatory Framework for Medical Devices" (2020) Proporciona una estructura modular para el desarrollo de marcos regulatorios progresivos en países de ingresos bajos y medios.
- "WHO Medical Device Technical Series" Serie de publicaciones sobre nomenclatura, evaluación de tecnología sanitaria, planificación de políticas y sistemas de gestión de calidad.
- "WHO Guidelines for Health Technology Assessment" (HTA) Promueve el uso de evidencia científica para la toma de decisiones sobre incorporación y priorización de productos sanitarios.
Sistema de precalificación (PQ) de productos sanitarios
La OMS ha establecido un Programa de Precalificación (WHO-PQ), inicialmente centrado en medicamentos esenciales, pero extendido a varios productos sanitarios considerados críticos para la salud pública. Este programa:
- Evalúa la calidad, seguridad y desempeño de productos como preservativos, esterilizadores, autoclaves, termómetros, dispositivos para malaria y VIH, entre otros.
- Incluye auditorías del fabricante, revisión del expediente técnico y ensayos de laboratorio.
- Sirve como requisito previo para la adquisición de productos por parte de agencias de Naciones Unidas (UNICEF, UNFPA, etc.).
- Este sistema es particularmente relevante en el contexto de emergencias sanitarias o compras masivas por programas internacionales.
Listas modelo y catálogos de productos sanitarios prioritarios
La OMS ha desarrollado herramientas de referencia para orientar la selección y adquisición de dispositivos médicos, entre ellas:
- Listado modelo de productos sanitarios esenciales por nivel de atención (2021). Incluye más de 200 productos organizados por nivel de complejidad (primaria, secundaria, terciaria).
- WHO Priority Medical Devices (PMD) for COVID-19 and Emergencies. Listas rápidas de productos indispensables para emergencias sanitarias.
- Compendio de innovaciones sanitarias. Promueve tecnologías adaptadas a contextos de bajos recursos.
Estas herramientas no solo orientan a gobiernos, sino también a donantes, ONGs y organismos multilaterales en compras y planificación de políticas.
Apoyo a la regulación nacional y regional
La OMS colabora estrechamente con autoridades regulatorias nacionales y regionales, especialmente en África, Asia y América Latina, mediante:
- Asistencia técnica directa para la redacción de leyes, reglamentos y procedimientos normativos.
- Capacitación en regulación basada en riesgo, evaluación clínica, inspección de fabricación, y vigilancia postcomercialización.
- Apoyo a la creación de autoridades regionales de regulación conjunta (ej. African Medicines Agency, Caribbean Regulatory System).
Esta función es esencial para reducir la fragmentación regulatoria y fortalecer la vigilancia de calidad en regiones vulnerables.
Normas de calidad y vinculación con otras entidades
Aunque la OMS no emite normas ISO, trabaja estrechamente con entidades como:
- ISO (Organización Internacional de Normalización): promueve la adopción de normas como ISO 13485 e ISO 14971 en países en desarrollo.
- IMDRF (Foro Internacional de Reguladores de Productos Sanitarios): la OMS actúa como observador y adopta sus principios como referencia técnica.
- Unidades de vigilancia: fortalece los sistemas de vigilancia postcomercialización para detectar fallos o productos defectuosos en circulación.
5.1.4 Tendencias actuales en la regulación de calidad
Evaluación del ciclo de vida completo del producto
Las nuevas regulaciones, como el MDR y las directrices de IMDRF, adoptan un enfoque de ciclo de vida que exige a los fabricantes considerar la calidad desde el diseño hasta la vigilancia postmercado.
Digitalización y productos sanitarios basados en software
El crecimiento del software como producto sanitario (SaMD) ha llevado a la creación de guías específicas, incluyendo requisitos de ciberseguridad, gestión del ciclo de vida del software y evaluación clínica basada en algoritmos.
Auditorías unificadas y programas de reconocimiento mutuo
Esfuerzos como el programa MDSAP (Medical Device Single Audit Program) permiten a los fabricantes someterse a una única auditoría que sea aceptada por múltiples autoridades regulatorias (Canadá, Australia, Brasil, Japón y EE.UU.), reduciendo la duplicación de esfuerzos y aumentando la eficiencia.
5.1.5 Implicaciones para los fabricantes y operadores económicos
Los fabricantes, representantes autorizados, importadores y distribuidores deben conocer y aplicar los requisitos normativos en cada mercado donde operan. Esto incluye:
- Mantener un sistema de gestión de calidad conforme a ISO 13485.
- Asegurar la documentación técnica y trazabilidad del producto.
- Establecer mecanismos de vigilancia y respuesta ante incidentes.
- Cumplir con las obligaciones éticas y legales en la comunicación con profesionales sanitarios y pacientes.
La calidad en los productos sanitarios no es un atributo accesorio, sino una exigencia central para su desarrollo, comercialización y uso clínico. La normativa internacional y las recomendaciones de organismos multilaterales proporcionan un marco coherente para garantizar dicha calidad a lo largo del ciclo de vida del producto. La armonización progresiva de los sistemas regulatorios y la convergencia hacia estándares comunes representan una oportunidad para mejorar la seguridad del paciente y facilitar el acceso a tecnologías sanitarias innovadoras en todo el mundo.
5.2 LA GESTIÓN DE LA CALIDAD EN LOS PRODUCTOS SANITARIOS: INTEGRACIÓN DE ISO 9001, ISO 13485 Y EL REGLAMENTO (UE) 2017/745
La calidad en los productos sanitarios no puede dejarse al azar. Para garantizar la seguridad del paciente, el desempeño clínico y el cumplimiento normativo, las organizaciones deben establecer sistemas de gestión de calidad (SGC) robustos y trazables. Las normas ISO 9001:2015 e ISO 13485:2016 proporcionan marcos internacionales ampliamente aceptados para la gestión de calidad. Sin embargo, en el espacio europeo, el Reglamento (UE) 2017/745 sobre productos sanitarios (MDR) impone requisitos legales vinculantes que deben ser plenamente integrados en el sistema de calidad de los fabricantes y operadores económicos. En adelante se relacionan estas tres referencias normativas y cómo se articulan para una gestión de calidad eficaz, conforme y orientada al ciclo de vida completo del producto sanitario.
5.2.1 Fundamentos de la gestión de calidad en productos sanitarios
La calidad en productos sanitarios implica la capacidad de cumplir de forma continua con los requisitos normativos, técnicos y clínicos aplicables. Esto exige una estructura organizativa sólida, procesos documentados, evidencia objetiva, y mecanismos eficaces de evaluación y mejora.
- La gestión de calidad se articula sobre tres pilares principales:
- Normas internacionales (ISO 9001 e ISO 13485).
- Requisitos legales vinculantes (MDR).
- Buenas prácticas de fabricación y vigilancia postcomercialización.
5.2.2 ISO 9001:2015 – Enfoque general para sistemas de calidad
Estructura y objetivos
ISO 9001 establece un modelo basado en procesos, centrado en la satisfacción del cliente y en la mejora continua. Aunque no está diseñada específicamente para productos sanitarios, sus principios fundamentales son útiles:
- Gestión del contexto de la organización.
- Liderazgo y compromiso.
- Gestión de riesgos y oportunidades.
- Seguimiento del desempeño y mejora.
Limitaciones en productos sanitarios
ISO 9001 no incluye requisitos clave del sector, como la gestión del riesgo clínico, la validación de procesos críticos, la trazabilidad del producto, ni la vigilancia postcomercialización. Por ello, aunque puede ser útil como base organizativa, no es suficiente para demostrar conformidad con el MDR.
5.2.3 ISO 13485:2016 – Norma específica para productos sanitarios
Características clave
ISO 13485 adapta los principios de ISO 9001 al sector sanitario, con un enfoque más prescriptivo y orientado al cumplimiento regulatorio. Sus características distintivas incluyen:
- Gestión del riesgo en todo el ciclo de vida (según ISO 14971).
- Trazabilidad reforzada, especialmente en productos implantables.
- Validación obligatoria de procesos especiales (esterilización, software).
- Control del entorno de trabajo y procesos de limpieza.
- Gestión documental y de registros clínicos.
- Acciones correctivas y preventivas, y vigilancia continua.
Ventajas
- Alineación con las expectativas de autoridades regulatorias (FDA, Health Canada, TGA, EMA).
- Reconocimiento en el programa MDSAP (Medical Device Single Audit Program).
- Base técnica para demostrar cumplimiento con el MDR en Europa.
5.2.4 EL REGLAMENTO (UE) 2017/745: Requisitos legales obligatorios
Principios generales
El Reglamento MDR establece las reglas para la comercialización, puesta en servicio y seguimiento postcomercialización de productos sanitarios en la UE. A diferencia de las normas ISO, el MDR es legislación obligatoria.
Incluye requisitos sobre:
- Clasificación del producto sanitario.
- Evaluación de la conformidad según clase de riesgo.
- Designación de persona responsable del cumplimiento normativo (PRRC).
- Documentación técnica (Anexo II).
- Investigación y evaluación clínica.
- Sistema UDI y trazabilidad.
Vigilancia postcomercialización y PMCF.
- Obligaciones para fabricantes, representantes autorizados, importadores y distribuidores.
Artículo 10: Obligaciones del fabricante
Uno de los artículos clave en materia de calidad es el Artículo 10 del MDR, que establece que:
“El fabricante establecerá, documentará, aplicará, mantendrá, actualizará y mejorará continuamente un sistema de gestión de calidad que garantice el cumplimiento del presente Reglamento de forma eficaz y proporcional a la clase de riesgo y tipo de producto fabricado”
El sistema debe abarcar:
- Estrategia para el cumplimiento normativo.
- Gestión del riesgo.
- Control del diseño y validación del producto.
- Procesos de producción y control.
- Vigilancia, PMCF (Seguimiento clínico postcomercialización) y seguimiento de tendencias.
- Mecanismos de mejora y CAPA (Gestión de no conformidades y acciones correctivas y preventivas).
Relación con ISO 13485
El MDR no exige explícitamente estar certificado en ISO 13485, pero el contenido de esta norma está alineado con los requisitos del Artículo 10, por lo que su implantación práctica es el modo más eficaz de cumplir legalmente.
5.2.5 Comparativa: ISO 9001, ISO 13485 y MDR
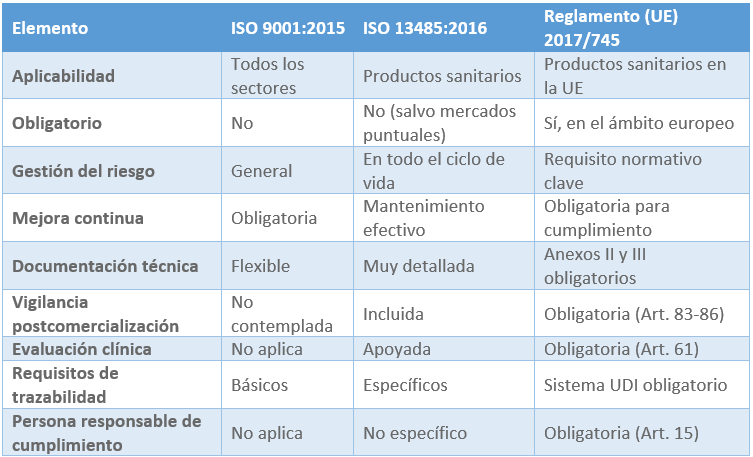
5.2.6 Implementación práctica: un sistema de calidad conforme a ISO 13485 Y MDR
- Diagnóstico de cumplimiento (gap analysis): comparación entre procesos actuales y requisitos del MDR + ISO 13485.
- Diseño del sistema: establecer procesos, roles y documentación.
- Gestión del riesgo clínico: implementación de ISO 14971 como base.
- Procedimientos de PMCF y vigilancia: definición de procesos para la recogida y análisis de datos del producto en uso real.
- Auditorías internas y revisión de la dirección: evaluación continua del sistema.
- Certificación y mantenimiento: obtención del certificado ISO 13485 por un organismo notificado y preparación para auditorías MDR.
La gestión de la calidad en productos sanitarios requiere la integración armónica de tres componentes:
- La estructura de procesos y mejora proporcionada por ISO 9001.
- La especificidad sectorial y enfoque regulatorio de ISO 13485.
- El cumplimiento legal obligatorio que impone el Reglamento (UE) 2017/745.
El uso combinado de estos marcos normativos permite a los fabricantes construir sistemas de calidad eficaces, conformes y sostenibles. En el actual contexto regulador europeo, la adopción de ISO 13485 como núcleo operativo del sistema de calidad se presenta no solo como una buena práctica, sino como una necesidad estratégica y legal.
5.3 MARCOS NORMATIVOS Y ESTÁNDARES DE CALIDAD EN PRODUCTOS SANITARIOS FUERA DE LA UNIÓN EUROPEA
La calidad de los productos sanitarios es una prioridad regulatoria a nivel global. Si bien la Unión Europea ha adoptado el Reglamento (UE) 2017/745 como marco legal integral, otros países y regiones también han desarrollado sistemas regulatorios robustos para asegurar la seguridad, eficacia y calidad de los productos sanitarios. Estos marcos comparten principios comunes —como la gestión del riesgo, la trazabilidad y la vigilancia postcomercialización— pero difieren en aspectos técnicos, estructurales y procedimentales.
A continuación, se resumen los principales requisitos y normas de calidad fuera de la UE, centrándose en Estados Unidos, Canadá, Japón, Australia, Brasil y las iniciativas globales del IMDRF.
5.3.1 REINO UNIDO: REGULACIÓN POST-BREXIT DE PRODUCTOS SANITARIOS
Contexto general
Tras su salida de la Unión Europea el 31 de enero de 2020, el Reino Unido (UK) dejó de estar sujeto al Reglamento (UE) 2017/745 (MDR) y al marco regulador comunitario. Desde entonces, el país ha iniciado la construcción de un sistema regulador propio para productos sanitarios, administrado por la Medicines and Healthcare products Regulatory Agency (MHRA).
Situación actual (transición regulatoria)
Durante el periodo de transición y hasta la implantación completa de la nueva normativa británica, el Reino Unido ha adoptado un sistema transitorio con las siguientes características:
- Continúa aceptando certificaciones CE hasta el 30 de junio de 2025 para la mayoría de productos sanitarios, lo que permite su comercialización temporal sin el marcado específico británico (UKCA).
- Exige registro obligatorio en la MHRA para todos los productos antes de ser comercializados en el mercado británico.
- A partir de julio de 2025, se requerirá el marcado UKCA (UK Conformity Assessed) para la mayoría de los productos sanitarios, emitido por un organismo de evaluación de conformidad británico (UK Approved Body).
Requisitos de calidad
En cuanto a los sistemas de gestión de calidad, la MHRA acepta y recomienda que los fabricantes:
- Apliquen un sistema basado en ISO 13485:2016, como base para demostrar conformidad con los requisitos esenciales del marco británico.
- Cumplan con los UK Medical Devices Regulations 2002 (en su versión enmendada), que incorporan elementos de la antigua Directiva 93/42/CEE y están siendo actualizados progresivamente hacia un modelo propio.
Nueva legislación en desarrollo
La MHRA publicó en 2022 su plan de reforma integral del marco regulador de productos sanitarios del Reino Unido, con los siguientes objetivos:
- Fortalecer la seguridad y vigilancia postcomercialización.
- Adoptar un modelo de evaluación clínica basado en evidencia real (real-world data).
- Establecer requisitos explícitos para software y productos digitales.
- Facilitar el acceso al mercado de productos innovadores.
Se espera que la nueva legislación entre en vigor a partir de 2025, con un enfoque similar al MDR europeo pero adaptado al contexto regulador británico.
Irlanda del Norte: una excepción particular
En virtud del Protocolo de Irlanda del Norte, este territorio sigue aplicando las normas del MDR y del IVDR de la Unión Europea para productos sanitarios. Por tanto:
- Los productos con marcado CE siguen siendo válidos en Irlanda del Norte.
- Los productos con marcado UKCA no pueden comercializarse en Irlanda del Norte.
- Se permite el uso de un "UKNI marking" en combinación con el CE cuando la evaluación de conformidad la haya realizado un organismo británico, pero este marcado no es válido para la UE.
5.3.2 ESTADOS UNIDOS – FDA Y EL 21 CFR PART 820
En Estados Unidos, la Food and Drug Administration (FDA) regula los productos sanitarios a través del Code of Federal Regulations (CFR), en particular: 21 CFR Part 820: establece el Quality System Regulation (QSR), es decir, los requisitos del sistema de calidad que los fabricantes deben cumplir.
Elementos clave del QSR
- Control del diseño y validación.
- Control de documentos y registros.
- Control de cambios.
- Validación de procesos y software.
- Gestión de proveedores.
- Acciones correctivas y preventivas (CAPA).
- Registros de producción y liberación.
Armonización con ISO 13485
La FDA ha anunciado una transición hacia la adopción del contenido de ISO 13485:2016 como base normativa, en un esfuerzo por reducir redundancias y facilitar el comercio internacional. El nuevo marco, llamado QMSR (Quality Management System Regulation), busca reemplazar la Parte 820 por una versión alineada con ISO 13485, respetando los principios de la legislación estadounidense.
5.3.3 CANADÁ – SISTEMA CMDR E ISO 13485
El organismo Health Canada regula los productos sanitarios bajo el Canadian Medical Devices Regulations (CMDR), incluido en la Ley de Alimentos y Medicamentos. La certificación es obligatoria
Para comercializar productos de Clase II o superior, los fabricantes deben estar certificados conforme a ISO 13485:2016, por un organismo reconocido por Health Canada. No se aceptan certificados ISO 9001.
Participación en MDSAP
Canadá es uno de los cinco países participantes en el Medical Device Single Audit Program (MDSAP), que permite a los fabricantes someterse a una auditoría única válida en múltiples jurisdicciones.
5.3.4 JAPÓN – PMDA Y J-GMP
En Japón, la Pharmaceuticals and Medical Devices Agency (PMDA) regula los dispositivos médicos bajo la Ley de Productos Farmacéuticos y Dispositivos Médicos (PMDL).
os fabricantes deben cumplir con las Japanese Good Manufacturing Practices (J-GMP), que integran elementos similares a ISO 13485 pero adaptados al marco normativo japonés.
- Evaluación del diseño (Design Review).
- Validación de procesos críticos.
- Control del entorno.
- Trazabilidad y gestión documental.
- Inspecciones por PMDA o autoridades delegadas.
Japón también acepta auditorías bajo MDSAP como parte de su proceso de autorización.
5.3.5 BRASIL – ANVISA Y RDC 665/2022
La Agência Nacional de Vigilância Sanitária (ANVISA) es la autoridad brasileña encargada de regular los productos sanitarios.
Desde 2022, rige la RDC 665/2022, que moderniza los requisitos del sistema de calidad para productos médicos, y se basa en los principios de:
- ISO 13485:2016.
- Buenas prácticas de fabricación específicas.
- Requisitos de documentación técnica y trazabilidad.
Brasil es también país miembro del programa MDSAP, y acepta las auditorías como parte de su sistema de registro.
5.3.6 AUSTRALIA – TGA Y CONFORMIDAD CON ISO 13485
La Therapeutic Goods Administration (TGA) regula los productos médicos en Australia bajo el Therapeutic Goods Act 1989.
Australia requiere que los fabricantes:
- Estén certificados según ISO 13485 por un organismo reconocido.
- Cumplan con el Australian Regulatory Guidelines for Medical Devices (ARGMD).
- Participen en auditorías MDSAP como vía aceptada para la evaluación de conformidad.
5.3.7 OTRAS JURISDICCIONES RELEVANTES
China – NMPA
- Exige auditorías de calidad a plantas de fabricación.
- Publicó en 2022 una normativa nacional alineada con ISO 13485.
- Fortalece los requisitos de evaluación clínica y trazabilidad.
Corea del Sur – MFDS
- Basado en ISO 13485 y GMP locales.
- Fuerte énfasis en validación de procesos y software.
India – CDSCO
- Marco aún en consolidación.
- Requiere cumplimiento con estándares BIS y componentes de ISO 13485.
5.3.8 INICIATIVAS GLOBALES: IMDRF Y ARMONIZACIÓN INTERNACIONAL
IMDRF – International Medical Device Regulators Forum
Este foro promueve la armonización de estándares y regulaciones a nivel global. Algunas de sus principales contribuciones incluyen:
- Documentos sobre clasificación de productos.
- Guías para la vigilancia postcomercialización.
- Buenas prácticas para software como producto sanitario (SaMD).
- Framework común para auditorías (base de MDSAP).
MDSAP – Medical Device Single Audit Program
Países participantes: EE.UU., Canadá, Brasil, Australia y Japón.
Beneficios:
- Auditoría única válida en múltiples países.
- Reducción de duplicidad de procesos.
- Mayor previsibilidad y eficiencia para fabricantes.
La calidad en los productos sanitarios es una prioridad compartida por los principales sistemas regulatorios del mundo. Aunque existen diferencias en los requisitos específicos, hay una clara tendencia hacia la convergencia en torno a ISO 13485:2016 como estándar común.
Conocer y aplicar los marcos regulatorios fuera de la UE —como los de la FDA, Health Canada, ANVISA, PMDA o TGA— es indispensable para garantizar el acceso global al mercado, cumplir con las exigencias normativas y proteger la salud de los pacientes en todos los contextos. La armonización internacional, impulsada por iniciativas como IMDRF y MDSAP, representa un paso firme hacia un sistema más coherente, eficiente y seguro a nivel global.
5.4 SISTEMA DE GESTIÓN DE LA CALIDAD SEGÚN ISO 13485: REQUISITOS Y APLICACIÓN EN EL SECTOR DE LOS PRODUCTOS SANITARIOS
La industria de los productos sanitarios constituye uno de los pilares fundamentales de los sistemas de salud modernos, al proporcionar herramientas y tecnologías indispensables para la prevención, diagnóstico, tratamiento y rehabilitación de enfermedades. Debido a su estrecha vinculación con la seguridad del paciente y la eficacia clínica, el desarrollo, fabricación y distribución de estos productos requiere un riguroso control de calidad y una sólida estructura regulatoria. En este contexto, la norma ISO 13485 se posiciona como el referente internacional para la implementación de sistemas de gestión de la calidad (SGC) específicos del sector.
A diferencia de otras normas de calidad de carácter general, la ISO 13485 fue diseñada expresamente para organizaciones involucradas en el ciclo de vida de los productos sanitarios. Su estructura y requisitos están enfocados en garantizar no solo la calidad del producto, sino también el cumplimiento normativo y la reducción de riesgos asociados al uso de los dispositivos médicos. Esta norma ha sido adoptada ampliamente en múltiples regiones del mundo, y en muchos casos constituye un requisito esencial para la comercialización de productos sanitarios en mercados altamente regulados como la Unión Europea, Canadá, Japón o Estados Unidos.
El enfoque de la ISO 13485 pone énfasis en la gestión del riesgo, la trazabilidad, la validación de procesos críticos y la mejora continua del sistema, aspectos clave en una industria donde los errores pueden tener consecuencias graves para la salud humana. Además, establece una base sólida para responder a requisitos reglamentarios específicos, como el Reglamento (UE) 2017/745 sobre productos sanitarios (MDR) o los requisitos del FDA 21 CFR Part 820.
El objetivo de este capítulo es analizar de forma exhaustiva los principales elementos de la norma ISO 13485, desde su estructura y principios generales hasta los requisitos técnicos más relevantes, incluyendo la gestión documental, la validación de procesos, la gestión de riesgos y la mejora del sistema. El enfoque adoptado será eminentemente técnico y práctico, con especial atención a su aplicación real en el entorno industrial y regulado, evitando referencias extensas a normas de carácter genérico como la ISO 9001.
5.4.1 Marco normativo y alcance de la ISO 13485
La norma ISO 13485:2016 especifica los requisitos para un sistema de gestión de la calidad cuando una organización necesita demostrar su capacidad para proporcionar productos sanitarios y servicios relacionados que cumplan de manera coherente con los requisitos del cliente y los requisitos reglamentarios aplicables. A diferencia de otros estándares genéricos, ISO 13485 está específicamente adaptada a la industria de productos sanitarios y su contexto altamente regulado.
5.4.1.1 Posición de la norma en el entorno regulatorio global
ISO 13485 no es, en sí misma, una ley o reglamento, pero su adopción es reconocida por múltiples autoridades sanitarias a nivel internacional como un marco adecuado para cumplir con los requisitos legales relativos al sistema de calidad. Es, por ejemplo:
- Aceptada por Health Canada como requisito para la obtención de la licencia de establecimiento.
- Reconocida por la FDA de Estados Unidos en su alineación progresiva con los requisitos del Quality System Regulation (QSR).
- Considerada en Europa como base para demostrar el cumplimiento parcial del sistema de calidad en el marco del Reglamento (UE) 2017/745 (MDR) y el Reglamento (UE) 2017/746 (IVDR).
En este contexto, la norma ISO 13485 se convierte en un puente entre los requisitos regulatorios y la implementación técnica de un sistema de gestión robusto y verificable.
5.4.1.2 Alcance organizativo y funcional
El alcance de la norma no se limita únicamente a los fabricantes de productos sanitarios. Pueden aplicarla también otros agentes económicos de la cadena de valor, incluyendo:
- Diseñadores y desarrolladores de productos sanitarios o software médico.
- Fabricantes por contrato y empresas de esterilización externa.
- Empresas de ensamblaje, embalaje o etiquetado.
- Distribuidores e importadores, especialmente cuando participan en actividades sujetas a control reglamentario.
- Organizaciones proveedoras de servicios asociados, como centros de mantenimiento o calibración de equipos médicos.
Este enfoque amplio permite adaptar el sistema de gestión de la calidad a funciones específicas, siempre que se declare claramente el alcance y se justifique la exclusión de requisitos no aplicables, como los relativos al diseño y desarrollo, si dicha actividad no se realiza.
5.4.1.3 Aplicación a lo largo del ciclo de vida del producto
ISO 13485 cubre de manera integral el ciclo de vida completo del producto sanitario, desde la concepción del diseño hasta el seguimiento postcomercialización. Esto incluye:
- Planificación y diseño del producto.
- Producción, control y liberación del producto.
- Embalaje, almacenamiento y distribución.
- Instalación y mantenimiento, si procede.
- Recolección de datos postcomercialización y retroalimentación.
Cada una de estas fases está sujeta a requisitos específicos que deben estar claramente definidos, documentados y controlados, con el fin de asegurar la conformidad.
5.4.2 Estructura de alto nivel de la norma
La ISO 13485:2016 adopta una estructura basada en cláusulas, alineada parcialmente con la estructura de alto nivel (HLS) de otras normas ISO, aunque con adaptaciones propias para ajustarse a los requisitos específicos del sector sanitario. Esta estructura facilita su implementación en organizaciones de distinto tamaño y complejidad, permitiendo integrar requisitos regulatorios con principios de gestión de la calidad.
5.4.2.1 Organización de la norma
La norma está dividida en ocho secciones, de las cuales las cláusulas 1 a 3 son introductorias (alcance, referencias normativas y términos) y las cláusulas 4 a 8 contienen los requisitos aplicables al sistema de gestión de la calidad:
- Cláusula 4 – Sistema de gestión de la calidad
- Cláusula 5 – Responsabilidad de la dirección
- Cláusula 6 – Gestión de los recursos
- Cláusula 7 – Realización del producto
- Cláusula 8 – Medición, análisis y mejora
Cada una de estas cláusulas se desarrolla a través de subapartados que incluyen requisitos detallados, muchos de los cuales están directamente relacionados con aspectos reglamentarios, como la trazabilidad, la validación de procesos especiales o la gestión del riesgo.
5.4.2.2 Enfoque basado en procesos
La norma promueve un enfoque basado en procesos, mediante el cual la organización debe identificar, planificar, implementar y controlar los procesos necesarios para el sistema de gestión de la calidad. Este enfoque permite:
- Asegurar la coherencia del producto con los requisitos especificados.
- Controlar las interacciones entre procesos.
- Evaluar el rendimiento de cada proceso y aplicar acciones correctivas cuando sea necesario.
El modelo de proceso también incluye mecanismos de retroalimentación y mejora continua, aunque la norma pone más énfasis en la gestión del riesgo que en el ciclo clásico de mejora PDCA (Plan-Do-Check-Act), como ocurre en otros estándares de calidad.
5.4.2.3 Responsabilidad de la dirección y compromiso organizativo
Una característica distintiva de la ISO 13485 es el rol activo que debe desempeñar la alta dirección en el establecimiento, implementación y mantenimiento del sistema de calidad. La dirección debe:
- Definir la política y los objetivos de calidad.
- Asignar responsabilidades específicas, incluida la designación de una persona responsable del cumplimiento de la regulación (en cumplimiento con MDR o MDSAP).
- Asegurar la disponibilidad de recursos y la competencia del personal.
- Realizar revisiones sistemáticas del sistema para garantizar su eficacia y adecuación.
Este enfoque refuerza la idea de que la calidad no es responsabilidad exclusiva del departamento técnico, sino un compromiso transversal que implica a toda la organización.
5.4.2.4 Gestión del riesgo como elemento estructural
A diferencia de otras normas, ISO 13485 exige explícitamente la aplicación del principio de gestión del riesgo a lo largo de todas las etapas del sistema de calidad. Este requisito no se limita al producto (como lo haría ISO 14971), sino que se extiende también a:
- La planificación de procesos.
- La selección y evaluación de proveedores.
- Las actividades de control del diseño y la producción.
- La gestión de cambios.
- Las acciones correctivas.
La gestión del riesgo debe estar documentada, actualizada y ser verificable. La integración de este enfoque fortalece la capacidad de la organización para prevenir fallos antes de que afecten al producto final o al usuario.
5.4.3 Requisitos clave del sistema de gestión de la calidad
Los requisitos establecidos en la norma ISO 13485 constituyen la base para la creación y mantenimiento de un sistema de gestión de la calidad eficaz, adaptable al sector de los productos sanitarios. Estos requisitos están organizados en torno a cinco grandes bloques funcionales que cubren desde la gestión documental hasta la mejora continua del sistema.
5.4.3.1 Requisitos generales y documentación
ISO 13485 exige que la organización establezca, documente, implemente y mantenga un sistema de gestión de la calidad, y que continuamente mejore su eficacia. Entre los elementos clave de este requisito se encuentran:
- Manual de calidad, que describa el alcance del sistema, los procedimientos documentados y la interacción entre procesos.
- Procedimientos obligatorios (por ejemplo, control de documentos, control de registros, acciones correctivas).
- Registros, que actúan como evidencia del cumplimiento y deben conservarse en condiciones adecuadas.
- Control de documentos y de registros, incluyendo aquellos en formato electrónico, garantizando su identificación, revisión, distribución y obsolescencia controlada.
La documentación debe reflejar la realidad operativa y no limitarse a un cumplimiento formal. Además, debe mantenerse actualizada y a disposición de las autoridades competentes cuando sea requerida.
5.4.3.2 Responsabilidad de la dirección
La alta dirección debe asumir un compromiso visible y verificable con el sistema de calidad, que se materializa a través de los siguientes elementos:
- Política de calidad, coherente con el propósito de la organización y enfocada a cumplir requisitos regulatorios.
- Objetivos de calidad, medibles y alineados con la mejora del producto y del sistema.
- Planificación del sistema de gestión, asegurando que los procesos y recursos están alineados con los objetivos.
- Revisión por la dirección, al menos una vez al año, para evaluar la eficacia del sistema, con base en datos de auditorías, reclamaciones, cambios, y desempeño general.
- Responsabilidad, autoridad y comunicación, incluyendo la designación de un responsable de calidad con autoridad definida para asegurar el cumplimiento de los requisitos.
Este apartado fortalece la gobernanza y la rendición de cuentas dentro de la organización.
5.4.3.3 Gestión de recursos
El éxito del sistema de calidad depende de que los recursos humanos, técnicos y materiales sean adecuados y estén gestionados correctamente:
- Personal competente, con formación, habilidades y experiencia apropiadas. Se deben mantener registros de la competencia y las acciones formativas.
- Infraestructura, incluyendo edificios, equipos y servicios asociados.
- Ambiente de trabajo, que puede incluir control de temperatura, humedad, contaminación o condiciones estériles, según la naturaleza del producto.
En este punto, se requiere que la organización analice las necesidades específicas de su proceso y producto, estableciendo controles apropiados.
5.4.3.4 Realización del producto
Esta cláusula constituye el núcleo operativo del sistema, ya que regula todas las actividades que llevan a la transformación de una necesidad del cliente en un producto sanitario seguro y conforme. Incluye:
- Planificación de la realización del producto, que debe estar documentada.
- Diseño y desarrollo, si es aplicable, incluyendo sus entradas, salidas, verificación, validación y control de cambios.
- Compras y control de proveedores, con evaluación basada en el impacto del proveedor sobre el producto final y en criterios definidos (calidad, cumplimiento, historial).
- Producción y prestación del servicio, incluyendo:
- Validación de procesos especiales (como esterilización o software).
- Control del producto no conforme en línea.
- Liberación del producto mediante criterios establecidos y personal autorizado.
- Trazabilidad, particularmente importante para productos implantables o activos, y para productos de Clase IIb y III en Europa.
- Manipulación, embalaje, almacenamiento y entrega, incluyendo el control de condiciones durante el transporte.
Esta sección establece un marco integral para controlar cada etapa que impacta sobre la calidad y seguridad del producto.
5.4.3.5 Medición, análisis y mejora
Una parte esencial del sistema de calidad consiste en evaluar el desempeño, identificar desviaciones y aplicar medidas correctivas o preventivas. La norma incluye:
- Seguimiento y medición del producto y procesos, con criterios definidos de aceptación.
- Auditorías internas, planificadas en función de la criticidad de los procesos y los resultados previos.
- Control del producto no conforme, con procedimientos para su identificación, segregación, evaluación y disposición, ya sea mediante retrabajo, rechazo o notificación al cliente.
- Análisis de datos, proveniente de múltiples fuentes: no conformidades, retroalimentación, auditorías, controles de proceso, etc.
- Acciones correctivas y preventivas, documentadas y basadas en causas raíz verificadas.
- Mejora continua, aunque la norma no exige explícitamente el uso del ciclo PDCA, sí requiere evidencia de esfuerzos sistemáticos para mantener y elevar la eficacia del sistema.
5.4.4 Gestión del riesgo y ciclo de vida del producto
La gestión del riesgo es uno de los pilares fundamentales de la norma ISO 13485 y, por extensión, del diseño, fabricación y distribución de productos sanitarios. A diferencia de otros sectores industriales donde el riesgo se enfoca principalmente en aspectos financieros o logísticos, en el ámbito sanitario los riesgos están directamente relacionados con la seguridad del paciente y la eficacia clínica del producto. Por ello, la norma establece una integración transversal de la gestión del riesgo en todas las etapas del sistema de calidad.
5.4.4.1 Enfoque normativo de la gestión del riesgo
La ISO 13485 exige que la organización aplique un enfoque basado en el riesgo a todos los procesos relacionados con el sistema de gestión de la calidad, no solo al diseño del producto. Este requisito representa una evolución frente a versiones anteriores, alineándose con regulaciones internacionales que demandan evidencia objetiva de la identificación, análisis, evaluación, control y seguimiento de riesgos.
Aunque la ISO 13485 no prescribe una metodología específica, sí hace referencia explícita a la norma ISO 14971 como el marco recomendado para la gestión del riesgo en productos sanitarios. Esta norma complementaria proporciona directrices sobre cómo:
- Identificar peligros asociados al producto.
- Estimar y evaluar los riesgos asociados.
- Controlar los riesgos mediante medidas adecuadas.
- Evaluar la aceptabilidad residual de los riesgos.
- Realizar seguimiento postcomercialización y revisión periódica.
5.4.4.2 Aplicación del riesgo en los procesos del sistema de calidad
A diferencia de una visión limitada al análisis de producto, ISO 13485 requiere aplicar el principio de gestión del riesgo a múltiples niveles del sistema de calidad. Algunos ejemplos concretos incluyen:
- Planificación del diseño: Evaluar el riesgo de fallos funcionales, errores de software o interacción con otros dispositivos.
- Selección de proveedores: Analizar el impacto del proveedor sobre la seguridad del producto, especialmente en componentes críticos o procesos tercerizados (como esterilización).
- Validación de procesos: Determinar riesgos asociados a la variabilidad de procesos no verificables por inspección (por ejemplo, sellado térmico, limpieza, dosificación).
- Gestión de cambios: Evaluar el impacto potencial de cambios en materiales, métodos, equipos o diseño sobre el desempeño y seguridad del producto.
- Gestión de las instalaciones: Identificar riesgos ambientales, de contaminación cruzada o de condiciones no controladas que puedan afectar la integridad del producto.
Este enfoque transversal refuerza la robustez del sistema y permite anticipar desviaciones antes de que se traduzcan en fallos del producto.
5.4.4.3 Documentación y trazabilidad del riesgo
La norma requiere que la gestión del riesgo esté documentada y trazable. Esto implica que la organización debe:
- Mantener registros del análisis de riesgos (listas de peligros, evaluaciones, matrices de riesgo, medidas de control).
- Asociar cada riesgo a una actividad o fase específica del producto o proceso.
- Justificar técnicamente la aceptabilidad del riesgo residual.
- Revisar los riesgos ante cualquier cambio significativo en el producto, el uso previsto o el entorno de aplicación.
- Actualizar los archivos de gestión del riesgo con la información de seguimiento postcomercialización (reclamaciones, alertas, incidentes).
En productos sujetos a evaluación de la conformidad con intervención de organismos notificados, estos documentos deben estar incluidos en el expediente técnico y disponibles para revisión.
5.4.4.4 Consideraciones específicas en el ciclo de vida del producto
La gestión del riesgo debe mantenerse activa durante todo el ciclo de vida del producto, desde su concepción hasta su retirada del mercado. Algunas implicaciones clave son:
- Durante el diseño, se analiza el uso previsto y los posibles usos indebidos.
- En la producción, se evalúan riesgos derivados del proceso y de la variabilidad de materiales o equipos.
- Tras la comercialización, se monitorean señales de campo para detectar riesgos no previstos.
- Si se detecta un riesgo no aceptable, se deben implementar medidas correctivas, que pueden incluir modificaciones técnicas, formación al usuario, cambios en el etiquetado o incluso retirada del mercado.
La integración coherente y dinámica de la gestión del riesgo constituye una de las herramientas más poderosas de la ISO 13485 para garantizar que los productos sanitarios sean seguros, eficaces y conformes durante todo su ciclo de vida.
5.4.5 Validación y trazabilidad
La validación de procesos y la trazabilidad de productos son elementos clave en la gestión de la calidad de productos sanitarios, especialmente por su impacto directo sobre la seguridad del paciente y el cumplimiento regulatorio. La norma ISO 13485 establece requisitos específicos y detallados para garantizar que los procesos críticos se controlen adecuadamente, y que los productos puedan ser rastreados eficazmente a lo largo de su ciclo de vida.
5.4.5.1 Validación de procesos
La norma requiere que la organización valide cualquier proceso de producción o prestación del servicio cuyo resultado no pueda verificarse completamente mediante inspección o ensayo posterior. En otras palabras, cuando la calidad del producto no puede asegurarse únicamente con controles finales, es obligatorio demostrar, de forma documentada, que el proceso es capaz de producir resultados conformes de manera consistente.
Ejemplos típicos de procesos que requieren validación:
- Esterilización (por vapor, óxido de etileno, radiación, etc.).
- Envasado estéril y sellado térmico.
- Moldeo por inyección o extrusión de polímeros críticos.
- Procesos de limpieza o descontaminación de dispositivos reutilizables.
- Software embebido en dispositivos médicos o software utilizado en producción y control.
Elementos clave de la validación:
- Criterios de aceptación definidos previamente.
- Estudios de capacidad del proceso (repetibilidad, reproducibilidad).
- Validación de equipos y métodos de medición asociados.
- Revalidaciones periódicas, especialmente ante cambios en materiales, equipos o parámetros.
- Registros documentados de todas las actividades de validación.
La validación no es un evento aislado, sino una parte integral del ciclo de control del proceso, que debe mantenerse actualizada y alineada con los datos del desempeño real.
5.4.5.2 Validación de software
ISO 13485 introduce requisitos específicos para la validación del software utilizado en el sistema de gestión de la calidad, en los procesos de producción y en el control del producto. Esto incluye tanto software comercial como herramientas desarrolladas internamente.
Casos en los que aplica:
- Sistemas informatizados de control documental.
- Software utilizado en etiquetado, inspección, o liberación de lotes.
- Aplicaciones de control de equipos automatizados.
- Hojas de cálculo críticas para decisiones regulatorias o técnicas.
La validación debe demostrar que el software cumple con el uso previsto, bajo condiciones normales y anormales, y que no introduce riesgos no aceptables para la calidad del producto o la conformidad del sistema.
5.4.5.3 Requisitos de trazabilidad
La trazabilidad permite conocer la historia, aplicación o localización de un producto o componente a través de identificaciones registradas. ISO 13485 exige establecer niveles apropiados de trazabilidad, en función del tipo de producto, su riesgo asociado y los requisitos regulatorios aplicables.
Requisitos generales:
- Trazabilidad de materias primas críticas, componentes y productos intermedios.
- Trazabilidad de lotes o números de serie, según lo exijan las regulaciones o la clase del producto.
- Registros de personas responsables de las actividades de liberación, fabricación o control.
Requisitos específicos:
- Para productos implantables, se exige la trazabilidad completa desde el proveedor hasta el paciente (y viceversa).
- En entornos regulados por MDR o MDSAP, se requieren mecanismos para facilitar el rastreo inverso en caso de alertas o retiradas.
Los registros de trazabilidad deben mantenerse durante períodos definidos, que en algunos casos pueden extenderse muchos años tras la vida útil del producto, según el marco regulatorio de cada país.
5.4.5.4 Control del etiquetado y de los productos terminados
Como parte del sistema de trazabilidad, la organización debe asegurar que:
- Los productos estén claramente identificados durante todas las etapas (recepción, producción, almacenamiento, distribución).
- Las etiquetas y el etiquetado estén controlados y verificados antes de su aplicación.
- Se mantengan registros que permitan rastrear cada unidad o lote hasta sus condiciones de fabricación y distribución.
Esto resulta especialmente relevante en situaciones de producto defectuoso, incidentes o retiradas del mercado.
La validación de procesos y la trazabilidad no solo son requisitos técnicos, sino garantías prácticas de seguridad, responsabilidad y conformidad normativa, especialmente en una industria donde los errores pueden traducirse en daños graves para los pacientes o consecuencias legales importantes para la organización.
5.4.6 Control del diseño y desarrollo
La cláusula relativa al diseño y desarrollo de productos sanitarios en la norma ISO 13485 es uno de los elementos más complejos y sensibles del sistema de gestión de la calidad, ya que establece las bases para garantizar que los productos cumplen con los requisitos de seguridad, eficacia, funcionalidad y conformidad normativa desde su concepción.
Aunque este requisito puede ser excluido si la organización no realiza diseño (por ejemplo, si sólo fabrica bajo especificaciones del cliente), cuando sí se lleva a cabo esta actividad, el sistema debe incluir un control riguroso, documentado y verificable de todo el proceso de diseño.
5.4.6.1 Planificación del diseño y desarrollo
La organización debe establecer un plan documentado para el diseño y desarrollo del producto sanitario. Este plan debe incluir:
- Etapas del diseño y criterios de revisión, verificación y validación.
- Responsabilidades y autoridades asignadas para cada fase.
- Interfaces entre departamentos técnicos, clínicos, regulatorios y de calidad.
- Gestión de recursos, documentación y cronogramas.
El plan puede modificarse a medida que avanza el proyecto, pero los cambios deben documentarse y justificarse. Este enfoque proporciona estructura y trazabilidad al proceso.
5.4.6.2 Entradas del diseño
Las entradas del diseño constituyen los requisitos técnicos, normativos, clínicos y de usuario que el producto debe cumplir. Estas entradas deben ser:
- Completas, claras, sin ambigüedades.
- Verificables o medibles.
- Derivadas de requisitos reglamentarios, normas técnicas, necesidades clínicas y del usuario final.
Ejemplos incluyen:
- Requisitos funcionales del producto.
- Requisitos de seguridad eléctrica, compatibilidad electromagnética, biocompatibilidad, etc.
- Requisitos de etiquetado, empaque, esterilización o almacenamiento.
5.4.6.3 Salidas del diseño
Las salidas del diseño deben proporcionar información suficiente para fabricar, inspeccionar y utilizar el producto, y deben:
- Cumplir con las entradas del diseño.
- Incluir especificaciones técnicas, materiales, dibujos, instrucciones de uso y requisitos de conservación.
- Asegurar la trazabilidad entre entradas y salidas.
- Incluir criterios de aceptación para producción y controles de calidad.
Las salidas deben estar verificadas y aprobadas antes de pasar a producción.
5.4.6.4 Revisión del diseño
Las revisiones son evaluaciones formales realizadas en puntos clave del proceso para verificar el avance y la coherencia del diseño. Deben incluir:
- Participación multidisciplinar (calidad, ingeniería, clínica, regulación).
- Identificación de problemas y propuestas de mejora.
- Registro de observaciones, decisiones y acciones.
Deben realizarse al menos en etapas críticas: definición de requisitos, diseño detallado, verificación, validación, y antes de la transferencia a producción.
5.4.6.5 Verificación del diseño
La verificación busca demostrar que las salidas del diseño cumplen con las entradas especificadas. Esto puede implicar:
- Simulaciones o cálculos técnicos.
- Ensayos de laboratorio.
- Comparaciones con diseños previos ya aprobados.
- Revisión técnica de los documentos del diseño.
Debe documentarse claramente el método, los resultados y la conclusión de conformidad.
5.4.6.6 Validación del diseño
La validación busca asegurar que el producto final cumple con las necesidades del usuario y el uso previsto, bajo condiciones reales o simuladas. Puede incluir:
- Ensayos de funcionalidad con prototipos o lotes piloto.
- Estudios clínicos o de usabilidad.
- Evaluación de factores humanos.
Se requiere una muestra representativa del producto, trazabilidad de las condiciones de validación, y justificación de la aceptabilidad de los resultados.
5.4.6.7 Control de cambios del diseño
Los cambios en el diseño deben:
- Estar documentados, evaluados y aprobados antes de su implementación.
- Considerar los impactos técnicos, normativos, clínicos y de calidad.
- Revisar y actualizar documentación relacionada (plan de diseño, registros de verificación y validación, especificaciones).
La trazabilidad de los cambios es esencial, especialmente si afectan productos en fase de fabricación o ya comercializados.
5.4.6.8 Transferencia a producción
La fase final consiste en transferir el diseño validado a la producción bajo condiciones controladas. La organización debe asegurarse de que:
- La documentación esté completa y disponible (planos, instrucciones, controles).
- Los procesos críticos estén validados.
- El personal esté formado adecuadamente.
- Los sistemas de control y liberación estén definidos.
Una transferencia deficiente puede comprometer el rendimiento del producto y anular los beneficios del desarrollo bien ejecutado.
El control del diseño y desarrollo según ISO 13485 permite a la organización demostrar la conformidad del producto desde su origen, facilitando la aprobación regulatoria, reduciendo fallos en el mercado y protegiendo la seguridad del paciente.
5.4.7 Producción, liberación y postproducción
Una vez que el diseño ha sido verificado, validado y transferido correctamente, el control sistemático de las actividades de producción, inspección, liberación y seguimiento postcomercialización se convierte en un componente esencial del sistema de gestión de la calidad. La norma ISO 13485 establece una serie de requisitos técnicos y organizativos para asegurar que cada producto sanitario fabricado sea seguro, conforme y trazable, incluso después de su distribución.
5.4.7.1 Control de la producción
La organización debe planificar y llevar a cabo la producción bajo condiciones controladas, lo que implica:
- Disponibilidad de instrucciones documentadas para procesos, equipos, materiales y criterios de aceptación.
- Uso de equipos adecuados y calibrados, verificados antes de su utilización.
- Validación de procesos especiales, como esterilización o soldadura, cuando el resultado no pueda verificarse completamente por inspección posterior.
- Gestión del ambiente de trabajo, especialmente cuando pueda influir sobre la calidad del producto (por ejemplo, en salas limpias o zonas de montaje estéril).
- Controles en línea y puntos críticos definidos, con registro de resultados.
Estas condiciones aseguran que la producción se realice de forma reproducible y bajo control estadístico o documental suficiente.
5.4.7.2 Liberación del producto
La liberación de cada lote o unidad de producto sólo puede realizarse cuando:
- Se hayan completado todas las actividades de producción e inspección requeridas.
- Se haya verificado que el producto cumple con las especificaciones técnicas y requisitos reglamentarios.
- Un personal autorizado haya dado su aprobación formal, dejando evidencia documentada de la liberación.
En productos estériles, implantables o activos, este control es especialmente crítico. Algunos mercados (como la UE) requieren incluso la participación de un organismo notificado en la evaluación de la conformidad antes del marcado CE.
5.4.7.3 Control del producto no conforme
Durante la producción, cualquier producto que no cumpla con los criterios establecidos debe:
- Ser identificado y segregado para evitar su uso o entrega no intencionada.
- Ser evaluado y dispuesto según procedimientos documentados: retrabajo, reparación, uso condicional, rechazo o destrucción.
- Incluir un registro de la no conformidad, su análisis de causa raíz y, si aplica, las acciones correctivas necesarias.
En el caso de productos ya liberados, el sistema debe permitir rastrear el lote y tomar medidas como notificaciones al cliente, investigaciones o retiradas del mercado.
5.4.7.4 Instalación y mantenimiento
Si la organización proporciona actividades de instalación o mantenimiento, éstas también deben estar controladas. Es necesario:
- Documentar los procedimientos técnicos.
- Formar adecuadamente al personal técnico.
- Mantener registros de cada actividad realizada (incluyendo ubicación, fecha, resultados y técnico responsable).
Estas actividades también deben incluir la gestión de riesgos, particularmente cuando pueden afectar la seguridad del paciente o el cumplimiento normativo.
5.4.7.5 Seguimiento pos comercialización y retroalimentación
El seguimiento pos comercialización es un componente crítico para asegurar que el producto continúa cumpliendo con los requisitos a lo largo del tiempo. La norma exige que la organización establezca procesos para:
- Recoger, analizar y responder a la retroalimentación de clientes, usuarios o distribuidores.
- Detectar tendencias o señales tempranas de posibles problemas.
- Investigar incidentes relacionados con la seguridad del paciente.
- Implementar acciones correctivas o preventivas, si corresponde.
Este sistema debe estar alineado con los requisitos reglamentarios de vigilancia, como los definidos por el MDR (UE) o la FDA (reportes de eventos adversos), y puede requerir interacciones con autoridades sanitarias.
5.4.7.6 Retirada de productos y acciones de campo
Cuando se identifica un problema en el mercado que afecta la seguridad, la organización debe estar preparada para:
- Ejecutar acciones correctivas de campo (actualización de instrucciones, correcciones técnicas, revisiones en sitio).
- Gestionar la retirada del producto si es necesario, notificando a clientes, distribuidores y autoridades.
- Documentar el proceso completo: causa, impacto, lotes afectados, medidas tomadas, comunicación y cierre del evento.
Un sistema de retirada eficaz requiere una trazabilidad robusta, comunicación clara y toma de decisiones basada en el análisis de riesgo.
Las actividades de producción, liberación y seguimiento postventa conforman el núcleo operativo que permite a la organización mantener la calidad y seguridad del producto incluso después de su comercialización, cumpliendo tanto con los requisitos de la norma como con las expectativas regulatorias y del mercado.
5.4.8 Auditorías y mejora continua
En un entorno altamente regulado como el de los productos sanitarios, la capacidad de una organización para verificarse a sí misma, aprender de sus errores y mejorar continuamente es fundamental. La norma ISO 13485 establece un marco estructurado para asegurar que el sistema de gestión de la calidad se mantenga eficaz, se adapte a los cambios y sea capaz de prevenir la recurrencia de fallos.
Este enfoque está centrado en tres pilares: auditorías internas, gestión de no conformidades y acciones de mejora basadas en datos objetivos.
5.4.8.1 Auditorías internas
La organización debe planificar, implementar y mantener un programa de auditorías internas que cubra todos los elementos del sistema de gestión de la calidad, incluyendo:
- Frecuencia basada en el riesgo y la criticidad de los procesos.
- Auditores competentes e independientes del área auditada.
- Criterios de auditoría definidos previamente, como procedimientos internos, la norma ISO 13485, y requisitos regulatorios aplicables.
- Resultados documentados que incluyan hallazgos, conformidades, no conformidades y oportunidades de mejora.
- Seguimiento de las acciones correctivas hasta su cierre y verificación de eficacia.
Las auditorías internas no deben verse como un trámite, sino como una herramienta poderosa de verificación y mejora. Además, constituyen una preparación estratégica para auditorías de organismos notificados, clientes o autoridades regulatorias.
5.4.8.2 Control de las no conformidades
La norma requiere procedimientos documentados para gestionar productos, procesos o sistemas que no cumplan con los requisitos establecidos. Las no conformidades pueden detectarse en producción, inspecciones, auditorías, quejas, devoluciones, o durante actividades de seguimiento postcomercialización.
La gestión incluye:
- Identificación y registro de la no conformidad.
- Evaluación de impacto sobre la calidad del producto y la seguridad del paciente.
- Contención inmediata, incluyendo segregación, retrabajo o retirada, si procede.
- Investigación de la causa raíz con métodos sistemáticos (por ejemplo, 5 porqués, diagrama de Ishikawa).
- Implementación y seguimiento de acciones correctivas, con evaluación de eficacia.
El objetivo es evitar la repetición de errores, no solo corregir sus consecuencias.
5.4.8.3 Análisis de datos y toma de decisiones
Una gestión eficaz de la calidad debe basarse en datos medibles y analizables. ISO 13485 establece que la organización debe recopilar y analizar información proveniente de múltiples fuentes:
- Resultados de auditorías.
- No conformidades y acciones correctivas.
- Retroalimentación de clientes y usuarios.
- Controles de calidad del producto y del proceso.
- Indicadores de desempeño clave (KPIs).
- Tendencias del mercado y cambios regulatorios.
Este análisis permite:
- Identificar tendencias negativas o repetitivas.
- Evaluar la eficacia de las acciones correctivas.
- Priorizar mejoras en función del riesgo y el impacto.
- Apoyar la toma de decisiones basada en evidencia.
5.4.8.4 Mejora del sistema de gestión de la calidad
Aunque ISO 13485 no impone explícitamente el modelo PDCA (Planificar-Hacer-Verificar-Actuar), sí requiere que la organización implemente acciones para mantener y mejorar la eficacia del sistema. Estas mejoras pueden derivarse de:
- Resultados de auditorías internas o externas.
- Quejas de clientes o incidentes de campo.
- Cambios en procesos, productos o tecnologías.
- Revisiones por la dirección.
- Indicadores de calidad y desempeño.
La mejora puede adoptar múltiples formas:
- Revisión y actualización de procedimientos.
- Automatización de procesos o digitalización.
- Capacitación del personal basada en hallazgos reales.
- Reducción del tiempo de ciclo, del retrabajo o de la tasa de fallos.
El compromiso con la mejora continua es también una señal de madurez del sistema de calidad y de alineación con los principios fundamentales del marco regulador de los productos sanitarios.
El ciclo de auditoría, análisis y mejora constituye una columna vertebral para la sostenibilidad del sistema de gestión, asegurando que la organización no solo cumpla con los requisitos, sino que los integre en su cultura operativa para anticiparse a los problemas, adaptarse al cambio y mejorar constantemente la calidad y seguridad del producto.
La norma ISO 13485 proporciona un marco estructurado, específico y robusto para la implementación de un sistema de gestión de la calidad en el sector de los productos sanitarios. Su enfoque centrado en la gestión del riesgo, la trazabilidad, la validación de procesos y el cumplimiento normativo la convierte en una herramienta esencial para fabricantes, proveedores y demás actores de la cadena de valor.

5.5 LA GESTIÓN DEL RIESGO EN LOS PRODUCTOS SANITARIOS SEGÚN LA NORMA ISO 14971:2019
La gestión del riesgo es un componente esencial en el diseño, fabricación y uso de productos sanitarios. En este sector, el riesgo no se limita a cuestiones técnicas o de desempeño, sino que se relaciona directamente con la seguridad del paciente, el personal sanitario y el entorno clínico. Las normas internacionales, especialmente ISO 14971:2019, establecen un marco sistemático para identificar, evaluar, controlar y monitorear los riesgos asociados a los productos sanitarios a lo largo de todo su ciclo de vida. Este capítulo analiza la gestión del riesgo desde el enfoque normativo ISO, su integración con sistemas de calidad como ISO 13485, y su papel clave en el cumplimiento regulatorio global.
5.5.1 Fundamentos de la gestión del riesgo en productos sanitarios
¿Qué es el riesgo?
En el contexto de productos sanitarios, riesgo se define como la combinación de la probabilidad de que ocurra un daño y la severidad de ese daño. Esta definición, presente en ISO 14971, implica que no basta con reducir la posibilidad de fallo: también debe considerarse el impacto clínico potencial.
Enfoque por ciclo de vida
La gestión del riesgo debe aplicarse de manera continua desde la fase de diseño hasta la retirada del producto, incluyendo:
- Desarrollo y validación.
- Producción y control de calidad.
- Comercialización y uso clínico.
- Vigilancia postcomercialización.
Este enfoque por ciclo de vida está en línea con el Reglamento (UE) 2017/745, que exige una gestión del riesgo como parte estructural del sistema de calidad del fabricante.
5.5.2 NORMA ISO 14971:2019 – Gestión del riesgo para productos sanitarios
La ISO 14971:2019 es la norma internacional de referencia para la gestión del riesgo en productos sanitarios. Proporciona un proceso estructurado y documentado para:
- Identificación de peligros.
- Estimación del riesgo asociado a cada peligro.
- Evaluación del riesgo aceptable.
- Control del riesgo.
- Evaluación de la efectividad del control.
- Análisis del riesgo residual.
- Supervisión de los riesgos en uso real.
Estructura del proceso
El proceso según ISO 14971 incluye:
- Análisis del riesgo: identificación de peligros, estimación de probabilidad y severidad.
- Evaluación del riesgo: comparación con criterios de aceptabilidad predefinidos.
- Control del riesgo: medidas de reducción mediante diseño, protección o información.
- Evaluación del riesgo residual: análisis de los riesgos que persisten tras los controles.
- Evaluación del beneficio-riesgo: necesaria cuando el riesgo residual excede el umbral aceptable.
- Información de producción y postproducción: vigilancia del rendimiento y nuevos riesgos.
5.5.3 ISO/TR 24971:2020 – Guía complementaria a ISO 14971
El informe técnico ISO/TR 24971:2020 proporciona orientación práctica para implementar ISO 14971. Aunque no es una norma certificable, aclara conceptos clave y ofrece recomendaciones detalladas para:
- Establecer criterios de aceptabilidad de riesgos.
- Documentar el razonamiento clínico y técnico.
- Gestionar riesgos de software médico y ciberseguridad.
- Aplicar la norma a diferentes tipos de productos (implantables, combinados, software).
- Integrar la gestión del riesgo en el sistema de calidad.
Esta guía es especialmente útil para fabricantes pequeños o nuevos en el sector, y también para organismos notificados durante auditorías de conformidad.
5.5.4 Integración de la gestión del riesgo en el sistema de calidad (ISO 13485)
La norma ISO 13485:2016 exige explícitamente la gestión del riesgo como parte integral del sistema de gestión de calidad:
- Cláusula 4.1.2(b): exige que el fabricante aplique un enfoque basado en el riesgo a todos los procesos del SGC.
- Cláusula 7.1: la planificación del producto debe considerar los riesgos relacionados con la seguridad del mismo.
- Cláusula 7.3: el diseño y desarrollo deben incluir actividades sistemáticas de gestión del riesgo.
- Cláusula 8: también exige una vigilancia postcomercialización que incluya reevaluación de riesgos.
Esto implica que la gestión del riesgo no debe limitarse a un documento aislado, sino estar integrada de forma transversal en todos los procesos clave: diseño, fabricación, control de cambios, vigilancia y mejora continua.
5.5.5 Técnicas y herramientas utilizadas en la gestión del riesgo
Técnicas comunes:
- FMEA (Failure Modes and Effects Analysis): útil para procesos y diseño de producto.
- FTA (Fault Tree Analysis): permite analizar eventos complejos a través de árboles lógicos.
- HAZOP (Hazard and Operability Analysis): técnica estructurada para identificar desviaciones en sistemas complejos.
- Análisis de beneficio-riesgo: herramienta obligatoria cuando los riesgos residuales no son despreciables.
Matrices de riesgo
Una práctica común es utilizar matrices de probabilidad vs. severidad para categorizar los riesgos y definir umbrales de aceptabilidad, que deben ser previamente definidos y justificados clínicamente.
5.5.6 Gestión del riesgo en la etapa postcomercialización
ISO 14971 y el MDR exigen que la gestión del riesgo continúe tras la comercialización del producto. Esto se traduce en:
- Revisión periódica de los riesgos conocidos.
- Identificación de nuevos peligros emergentes.
- Actualización del análisis de riesgos en base a datos reales (real-world evidence).
- Integración con los sistemas de vigilancia y PMCF (Post-Market Clinical Follow-up).
Esto refuerza el enfoque dinámico y cíclico del riesgo: el análisis no termina tras la liberación del producto, sino que se retroalimenta continuamente.
5.5.7 Conexión con el reglamento (UE) 2017/745 y otros marcos regulatorios
El MDR integra la gestión del riesgo como uno de sus ejes fundamentales:
- Artículo 10.2: exige un sistema de gestión del riesgo durante todo el ciclo de vida.
- Anexo I: define requisitos generales de seguridad y desempeño basados en principios de gestión del riesgo.
- Evaluación clínica: debe tener en cuenta el análisis beneficio-riesgo.
En otros mercados (EE.UU., Canadá, Japón, Brasil), los principios de ISO 14971 también están incorporados en sus marcos normativos o se exigen como parte de las auditorías regulatorias (por ejemplo, MDSAP).
5.5.8 Caso práctico: aplicación de la gestión del riesgo en una bomba de infusión volumétrica
Descripción del producto
Bomba de infusión volumétrica electrónica de uso hospitalario, Clase IIb según MDR. Administra soluciones intravenosas con precisión programable (medicación, fluidoterapia, nutrición parenteral). Uso en adultos, pediátricos y neonatales bajo supervisión profesional.
1. Identificación de peligros
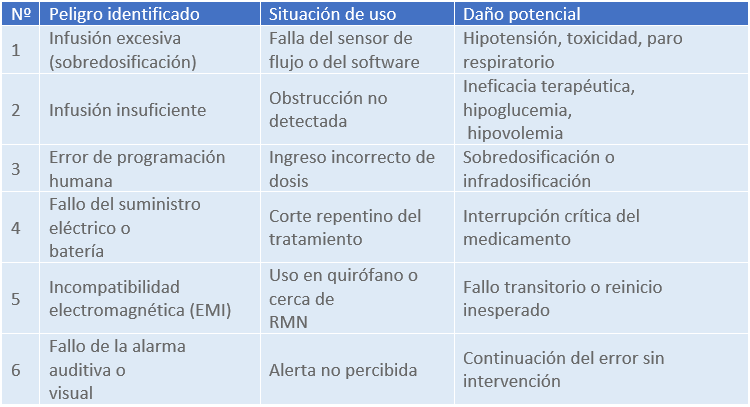
2. Estimación del riesgo inicial
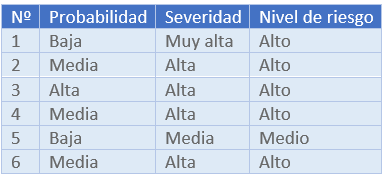
3. Medidas de control del riesgo
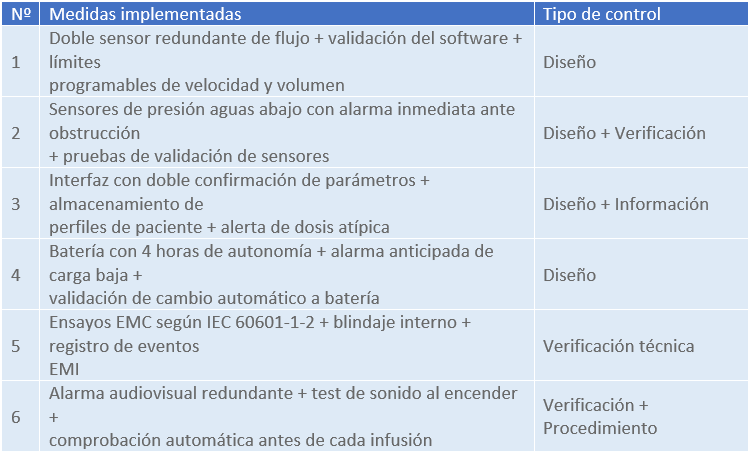
4. Evaluación del riesgo residual
5. Actividades de postproducción
- Integración con el sistema de vigilancia postcomercialización (PMS).
- Registro y análisis de quejas por alarmas no detectadas o errores de programación.
- Revisión anual del análisis de riesgos como parte del reporte de seguridad clínica.
- Actualización de firmware para mejorar algoritmos de validación de parámetros.
- Incorporación del producto en el plan de seguimiento clínico posterior a la comercialización (PMCF) para evaluar exactitud y fiabilidad en entorno real.
Conclusión del caso
Este ejemplo muestra cómo aplicar los principios de ISO 14971:2019 en un dispositivo crítico como una bomba de infusión. Destacan las siguientes buenas prácticas:
- Uso de controles múltiples (redundancia, validación y alarmas).
- Consideración del error humano como fuente de riesgo.
- Integración con los sistemas de calidad (ISO 13485) y vigilancia postcomercialización.
- Evaluación objetiva del riesgo residual frente al beneficio clínico esperado.
La trazabilidad y documentación de todo el proceso permiten demostrar conformidad normativa, facilitar auditorías regulatorias y, sobre todo, mejorar la seguridad del paciente en el entorno clínico.
FMEA - Bomba de Infusión

Matriz resumen FMEA:
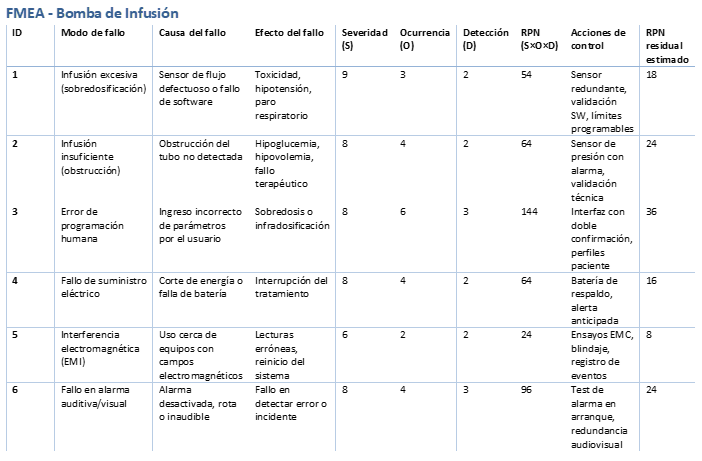
La gestión del riesgo es una función estratégica en el desarrollo y control de productos sanitarios. Las normas ISO 14971:2019 e ISO/TR 24971:2020 proporcionan una guía completa para implementar un proceso estructurado, trazable y alineado con los requisitos regulatorios internacionales. Su integración en el sistema de calidad según ISO 13485:2016 es indispensable para garantizar la seguridad del paciente, la eficacia del producto y la sostenibilidad del cumplimiento normativo.
En un entorno donde la innovación tecnológica avanza rápidamente y las expectativas regulatorias aumentan, una gestión del riesgo madura y proactiva se convierte en un verdadero motor de calidad, confianza y competitividad.
5.6 LAS NORMAS UNE
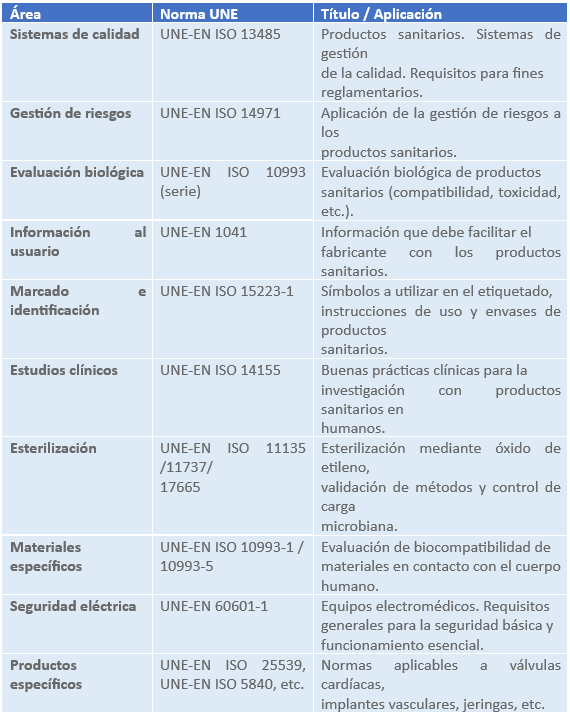
Las normas UNE (acrónimo de Una Norma Española) desempeñan un papel esencial en el sector de los productos sanitarios como referencia técnica para garantizar la seguridad, el rendimiento y la calidad de los dispositivos comercializados en España y en el entorno europeo. Estas normas son elaboradas, adoptadas o traducidas por la Asociación Española de Normalización (UNE) y, en muchos casos, corresponden a versiones nacionales de normas armonizadas europeas (EN) o internacionales (ISO o IEC). Aunque las normas UNE no son de cumplimiento obligatorio per se, su aplicación ofrece una presunción de conformidad con los requisitos esenciales establecidos en el Reglamento (UE) 2017/745 sobre productos sanitarios y el Reglamento (UE) 2017/746 sobre productos para diagnóstico in vitro, siempre que estén citadas en el Diario Oficial de la Unión Europea como normas armonizadas. En la práctica, su uso facilita a los fabricantes la elaboración de la documentación técnica, la evaluación de riesgos, la validación de procesos y el diseño de ensayos clínicos, entre otros aspectos críticos del ciclo de vida del producto. Además, su inclusión en los procedimientos internos de calidad —por ejemplo, dentro de un sistema basado en la norma ISO 13485— refuerza el cumplimiento normativo frente a auditorías e inspecciones regulatorias. De este modo, las normas UNE constituyen una herramienta clave para alinear los procesos técnicos y de fabricación con las exigencias legales y regulatorias aplicables a los productos sanitarios.
Ejemplos de normas UNE aplicables a productos sanitarios
Para obtener más información sobre las normas que regulan la fabricación de los productos sanitarios, se puede consultar el documento realizado por la Asociación Española de Normalización (UNE) donde se resumen las normas disponibles, así como información de como se elaboran por los comités de expertos. El enlace es:
https://www.une.org/normalizacion_documentos/Apoyo a la sanidad.pdf
5.7 LA EVALUACIÓN BIOLÓGICA DE PRODUCTOS SANITARIOS MEDIANTE LA NORMA UNE-EN ISO 10993-1:2021
La norma UNE-EN ISO 10993 es una serie de normas internacionales que establece los principios y métodos para la evaluación biológica de productos sanitarios. Su propósito principal es garantizar la seguridad biológica de los materiales utilizados en dispositivos médicos antes de su comercialización, evaluando su compatibilidad con el cuerpo humano.
Esta norma forma parte esencial del marco regulador internacional en materia de productos sanitarios y es complementaria a los requisitos generales de seguridad y funcionamiento definidos por reglamentos como el Reglamento (UE) 2017/745. La serie UNE-EN ISO 10993 consta de múltiples partes, cada una dedicada a un aspecto específico de la evaluación biológica, siendo la parte 1 la de carácter general y directivo.
Parte 1: Evaluación y ensayo en un proceso de gestión de riesgos
La UNE-EN ISO 10993-1 establece los principios generales para planificar una estrategia de evaluación biológica integrada en el sistema de gestión de riesgos del producto. Esta parte determina qué ensayos son necesarios en función de factores como:
- La naturaleza del dispositivo.
- El tipo y duración del contacto con el cuerpo.
- Las características fisicoquímicas de los materiales.
El enfoque de la norma prioriza el uso de información ya existente, la evaluación química y toxicológica de los materiales, y la reducción del uso de ensayos in vivo, alineándose con el principio de las 3R (reducir, reemplazar, refinar) en la experimentación animal.
Otras partes relevantes de la norma
Algunas de las partes más significativas de la serie incluyen:
- Parte 3: Ensayos de genotoxicidad, carcinogenicidad y toxicidad reproductiva.
- Parte 5: Ensayo de citotoxicidad in vitro.
- Parte 10: Ensayos de irritación y sensibilización.
- Parte 11: Ensayos de toxicidad sistémica.
- Parte 17: Umbrales de toxicidad y análisis de riesgo toxicológico.
Aplicación práctica
La aplicación de la norma UNE-EN ISO 10993 implica una evaluación científica estructurada, la recopilación de datos fisicoquímicos y toxicológicos de los materiales, la realización de pruebas biológicas donde sea necesario, y la documentación de todo el proceso en un informe de evaluación biológica. Esta documentación debe estar disponible durante el proceso de evaluación de la conformidad del producto sanitario
5.8 PREVENCIÓN DE RIESGOS POR OBJETOS PUNZANTES Y CORTANTES
La normativa europea y española sobre prevención de riesgos por objetos punzantes y cortantes en el sector sanitario se basa en una directiva europea específica que ha sido transpuesta al ordenamiento jurídico español mediante un Real Decreto.
Normativa Europea:
Directiva 2010/32/UE del Consejo, de 10 de mayo de 2010
Esta directiva aplica el Acuerdo Marco europeo firmado por los interlocutores sociales del sector hospitalario (HOSPEEM y EPSU) sobre la prevención de lesiones causadas por objetos punzantes o cortantes en el entorno hospitalario y sanitario
- Tiene por objetivo proteger a los trabajadores sanitarios frente a riesgos de infecciones causadas por pinchazos accidentales.
- Establece medidas preventivas y organizativas: formación, procedimientos seguros, participación del personal, uso de dispositivos de bioseguridad.
- Obliga a implantar dispositivos médicos con protección contra pinchazos, cuando sea técnicamente posible.
Normativa Española:
Se regula mediante el Real Decreto 598/2015, de 3 de julio, que transpone la Directiva 2010/32/UE al ordenamiento jurídico español.
Añade un nuevo capítulo al RD 664/1997 (Capítulo VI: Lesiones por objetos punzantes), por el que obliga a adoptar medidas para evitar lesiones con agujas, bisturíes, lancetas y otros instrumentos afilados.
Refuerza la obligación de:
- Aplicar procedimientos seguros.
- Proporcionar formación específica.
- Evaluar riesgos específicos en cada puesto.
- Eliminar prácticas peligrosas como el reencapuchado de agujas.
- Promover el uso de materiales con dispositivos de seguridad integrados.
Ámbito Norma Contenido principal
UE Directiva 2010/32/UE Protección frente a lesiones por objetos cortopunzantes en el sector sanitario
España Real Decreto 664/1997 modificado por RD 598/2015 Obligación de adoptar medidas técnicas y organizativas específicas para prevenir pinchazos accidentales
5.9 PRODUCTOS SANITARIOS CON DOBLE CERTIFICACIÓN MDR Y EPI
En determinados contextos, existen productos sanitarios que, además de estar destinados a fines médicos, también cumplen funciones de protección del usuario frente a riesgos físicos, químicos o biológicos. En estos casos, dichos productos están sujetos a una doble regulación, conforme al Reglamento (UE) 2017/745 sobre productos sanitarios (MDR) y al Reglamento (UE) 2016/425 sobre Equipos de Protección Individual (EPI).
La doble certificación es aplicable cuando un producto:
- Tiene una finalidad médica, como diagnóstico, prevención o tratamiento de enfermedades (MDR),
- Proporciona protección al usuario frente a riesgos para su salud o seguridad (EPI).
Este solapamiento se produce principalmente en productos como:
- Mascarillas médicas tipo IIR, que protegen tanto al paciente como al profesional frente a fluidos.
- Guantes de examen o quirúrgicos, con funciones médicas y de barrera frente a agentes infecciosos.
- Pantallas faciales o gafas de protección, si se utilizan con fines sanitarios y de protección individual.
Para estos productos, los fabricantes deben:
- Asegurar el cumplimiento simultáneo de los requisitos esenciales de ambos reglamentos.
- Realizar una evaluación de conformidad doble, incluyendo ensayos normalizados (por ejemplo, EN 14683 para mascarillas y EN 149 o EN 374 para protección respiratoria o química).
- Contar con un organismo notificado competente en ambas normativas para la certificación del producto.
- Emitir una declaración de conformidad que indique explícitamente el cumplimiento de los dos reglamentos.
La Comisión Europea ha publicado orientaciones (por ejemplo, la guía MDCG 2021-27) para ayudar a los fabricantes y organismos notificados a aplicar correctamente la doble regulación.
Por ello, los productos sanitarios con función de protección individual deben cumplir un doble marco reglamentario, lo que implica una gestión técnica y documental rigurosa para garantizar su seguridad, eficacia clínica y capacidad de protección frente a riesgos laborales.
BIBLIOGRAFÍA
- https://es.linkedin.com/pulse/el-instrumental-quir%C3%BArgico-algo-de-su-historia-albarracin-miranda.
- historia.nationalgeographic.com.es/a/sofisticacion-antiguo-egipto-protesis-hace-3000-anos_11639
- portalcomunicacion.uah.es/diario-digital/actualidad/medicina-medieval-una-etapa-oscura-pero-llena-de-supersticiones/
- www.salusplay.com/apuntes/quirofano-y-anestesia/tema-3-instrumentacion
- wfsahq.org/es/about/history/history-of-anaesthesia
- www.bbvaopenmind.com/ciencia/investigacion/joseph-lister-el-hombre-que-esterilizo-la-cirugia/
- www.revistamedicinaycultura.fmposgrado.unam.mx/index.php/2023/01/24/joseph-lister-biografia/
- identyd.com/historia-autoclave/
- www.elsevier.es/es-revista-revista-argentina-radiologia-383-articulo-wilhelm-conrad-roentgen-el-descubrimiento-S0048761916301545
- www.fda.gov/medical-devices/overview-device-regulation/history-medical-device-regulation-oversight-united-states
- formasefh.sefh.es/tecnifarmh/curso-productos-sanitarios/curso-productos-sanitarios.pdf
- www.aemps.gob.es/la-aemps/legislacion/legislacion-sobre-productos-sanitarios/
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- www.aemps.gob.es/productos-sanitarios/investigacionclinica-productossanitarios/
- UNE-EN ISO 14971:2020 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN 62366-1:2015/A1:2020 (Ratificada) Productos sanitarios. Parte 1: Aplicación de la ingeniería de usabilidad a los productos sanitarios.
- Aplicación de la gestión de riesgos a los productos sanitarios (une.org)
- 231219_GuiaValidacion_Tec_Sanitarias-1.pdf (inndromeda.es)
- Ciclo de vida de producto sanitario archivos - Consultoría de producto sanitario
- Ciclo de vida de producto sanitario y gestión de riesgos - Fernando Atienza
- Producto sanitario definición, características y usos (productossanitarios.net)
- Risk Management in the Medical Device Industry (dqsglobal.com)
- Design Thinking en Producto Sanitario ¿Qué es idear, prototipar y testar? (ambit-bst.com)
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Ley 29/2006, de 26 de julio de garantías y uso racional del medicamento.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS): https://www.aemps.gob.es/
- Federación Española de Empresas de Tecnología Sanitaria (FENIN): https://www.fenin.es/
- Ministerio de Ciencia e Innovación (PERTE Salud de Vanguardia): https://www.ciencia.gob.es/
- European Commission. (2021). Guidance on the application of the MDR and IVDR during the COVID-19 pandemic. Publications Office of the European Union. https://health.ec.europa.eu/system/files/2021-05/md_guidance_md_011_en_0.pdf
- Ghisellini, P., Cialani, C., & Ulgiati, S. (2016). A review on circular economy: The expected transition to a balanced interplay of environmental and economic systems. Journal of Cleaner Production, 114, 11-32. https://doi.org/10.1016/j.jclepro.2015.09.007
- Organización Internacional del Trabajo (OIT). (2023). Declaración de principios y derechos fundamentales en el trabajo. https://www.ilo.org/global/lang--es/index.htm
- Organización Mundial de la Salud (OMS). (2022). Health product policy and standards. https://www.who.int/teams/health-product-and-policy-standards
- Parlamento Europeo y Consejo de la Unión Europea. (2017). Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo de 5 de abril de 2017 sobre los productos sanitarios. Diario Oficial de la Unión Europea. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX%3A32017R0745
- Porter, M. E., & Kramer, M. R. (2011). Creating shared value. Harvard Business Review, 89(1/2), 62-77.
- OMS. (2015). Ética de la salud mundial: cuestiones clave.
- Declaración de Helsinki de la AMM – Principios éticos para las investigaciones médicas con participantes humanos – WMA – The World Medical Association.
- Global health ethics: key issues (who.int)
- https://lac.saludsindanio.org/cambio-climatico-y-salud/huella-climatica-del-sector-salud
- https://www.miteco.gob.es/content/dam/miteco/es/cambio-climatico/temas/mitigacion-politicas-y-medidas/docuementoexplicativosello_tcm30-479000.pdf
- https://accionclimaticaensalud.org/sites/default/files/2021-06/huellaclimatica.pdf
- Estrategia de Economía Circular de Euskadi 2030 - Prevención de la contaminación, inspección y control ambiental - Euskadi.eus
- Residuos Sanitarios (miteco.gob.es)
- Guía de procedimientos de esterilización en el medio hospitalario. file:///C:/Users/50820026P/Documents/OneDrive%20-%20Madrid%20Digital/PERSONAL/MASTER%20ordenador%20sollube/2.%20TEMARIO%20MASTER%20FARMA%20Y%20PS/procedementos_esterilizacion.pdf
- Curso básico de Gestion de Productos Sanitarios ANECORM
- https://www.aemps.gob.es/informa/la-aemps-pone-en-marcha-una-nueva-aplicacion-para-la-comunicacion-de-fabricacion-de-productos-sanitarios-in-house-por-hospitales/
- REGLAMENTO (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) n.o 178/ 2002 y el Reglamento (CE) n.o 1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo (boe.es)
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- UNE-EN ISO 9001:2015. Sistemas de gestión de la calidad. Requisitos (ISO 9001:2015)
- UNE-EN ISO 13485:2018 Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios
- UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN ISO 10993-1:2021 Evaluación biológica de productos sanitarios.
- Directiva 2010/32/UE Protección frente a lesiones por objetos cortopunzantes en el sector sanitario
- Real Decreto 664/1997 modificado por RD 598/2015 Obligación de adoptar medidas técnicas y
- Organizativas específicas para prevenir pinchazos accidentales