La esterilización es un proceso que elimina o destruye por completo todas las formas de vida microbiana, incluidas bacterias, virus, hongos y esporas, presentes en el producto con el objetivo es garantizar que el producto esté libre de microorganismos viables y se mantenga estéril hasta su utilización haciendo seguro su uso en procedimientos médicos o quirúrgicos según las regulaciones sanitarias.
Para conseguir una buena esterilización se debe tener en cuenta todo un proceso, no solo el acto de la esterilización del producto. Previamente a la fase de esterilización se debe realizar la descontaminación y limpieza del producto y el empaquetado, en ese momento se podrá realizar el acto de esterilizar y posteriormente se tendrá que tener cuidado con el almacenaje y distribución para no dañar el empaquetado y que el producto llegue al usuario en perfectas condiciones.
Existen diversos métodos de esterilización con sus correspondientes validaciones y requisitos normativos, siempre con un sistema de trazabilidad para poder detectar cualquier fallo. Se debe tener especial cuidado en el control de la esterilización para evitar riesgos como la infección nosocomial que podría largar la estancia hospitalaria con el consiguiente sufrimiento al paciente y un aumento del gasto.
6.1 PROCESO DE ESTERILIZACIÓN
En una central de esterilización debemos disponer de diferentes áreas claramente diferenciadas y con los flujos de trabajo bien definidos.
En primer lugar, nos encontraremos con el área de descontaminación y limpieza. Aquí se debe realizar una limpieza exhaustiva del material. La limpieza se debe realizar lo antes posible tras el uso y en el caso de retrasarse se debe dejar el material que lo permita a remojo o en una solución pre-clean especifica que retrase la solidificación de sangre y otros restos.
Se emplea procedimientos físico químico con detergente enzimático y agua a una temperatura que depende de las instrucciones del detergente, para conseguir que el detergente haga su efecto y evitar la coagulación de las proteínas (albuminas) que dificultaría la limpieza, pudiendo ayudarse en una primera fase con pistolas de presión o cepillado si el material lo permitiera y fuera necesario, para eliminar cualquier resto de materia orgánica y suciedad.
Para la limpieza, el material que lo permita debe ser desmontado en todas sus piezas y el instrumental debe estar siempre abierto.
Actualmente los métodos utilizados para limpiar el material son tres:
- Limpieza manual. Existen materiales muy delicados que el lavado se puede realizar con agua y detergente, pero se debe realizar con esmero y cuidado como pueden ser las pinzas de oftalmología para no dañarlas. También tenemos material que no puede ser mojados como cables de luz fría, ópticas o motores estos materiales no deben ser sumergidos. Debemos recordar que el material canulado debe ser lavado también por el interior.
- Limpieza automática. Se realiza en aparato automático que recuerda a un lavavajillas, con diferentes tipos de gradillas e incluso tubos para poder colocar el material canulado largo, tipo tubos de endoscopia, insertado para un lavado adecuado del interior. El material se debe ubicar separado para que se pueda realizar una buena circulación de los detergentes y el agua durante el lavado. Este tipo de lavados nos permite realizar trazabilidad, debemos colocar un control de proteínas secas para asegurarnos que el lavado se ha realizado correctamente. Es el método adecuado para el material termosensible. Existen diferentes programas, los más utilizados son para instrumental, delicado, para material canulado y rápido.
- Limpieza con ultrasonidos. Es un método que funciona por ondas longitudinales producidas en el líquido donde está el material sumergido. El material se coloca en una cubeta con el agua y detergente y se deja actuar durante periodos de 15 minutos, después de cada periodo se va revisando si el material está limpio. Al terminar se debe realizar un aclarado para eliminar todos los restos. Este método es adecuado para material de acero inoxidable nunca para plásticos y gomas.
Después del lavado es muy importante realizar un riguroso secado, en el método automatico es parte del ciclo, y lubricado de las piezas que lo necesiten. El secado se realizará con paños que no desprendan hilos, ni pelusas y las partes canuladas con pistolas de aire.
La segunda zona es la de empaquetado. Antes de empaquetar se debe realizar una revisión de todas las piezas para comprobar que funcionan correctamente, que no se han despuntado ni estropeado en el lavado.
El objetivo del empaquetado es mantener el material estéril hasta su utilización. Es un procedimiento en el que cobra especial importancia la selección del material de envoltura y una técnica que garantice la integridad del paquete antes y durante su procesamiento, hasta el momento de uso.
El paquete debe preservar la esterilidad de su contenido, por lo que utilizar doble envoltura es la opción que proporciona una mayor seguridad. Los objetos deben estar empaquetados de tal forma que el envoltorio que los contiene pueda ser abierto y su contenido extraído sin contaminaciones y con la máxima facilidad para el usuario, la mejor manera es con doble pestaña. Para ello, se debe elegir el mejor material según el método de esterilización y el material que se empaqueta. Tiene que presentar las siguientes características: ser compatible con el método de esterilización permitiendo la penetración y eliminación del agente esterilizador, permitir precinto e identificación del material y garantizar la resistencia e integridad del paquete para hacer de barrera al material esterilizado.
Los materiales utilizados para el empaquetado deben llevar el marcado CE de clase I, actualmente son papel crepado de celulosa, tejido sin tejer de celulosa y poliéster, polipropileno y bolsas y rollos mixtos y contenedores. El textil cada vez es menos utilizado. Según la esterilización los empaquetados serian:
- Empaquetado para esterilización por vapor (Autoclave)
- Material individual: doble bolsa mixta, colocadas en el mismo sentido. Se utilizan normalmente bolsas mixtas con papel poroso y, si el material es pesado o con alto riesgo de rotura, en bolsa mixta con tejido sin tejer. Las bolsas tienen una cinta indicadora externa, como control químico del proceso, y se cierran con selladora térmica.
- Cajas perforadas: se realizará un empaquetado de barrera con bolsa mixta de tejido sin tejer o con envoltura de papel crepado o de tejido sin tejer, cerrando la envoltura con cinta adhesiva indicadora del proceso. En su interior siempre se colocará un indicador químico.
- Contenedor con filtro o válvula: llegará cerrado a la Central de Esterilización y con un indicador químico en su interior. Siempre se colocará en el exterior una etiqueta con indicador químico de proceso.
- Para asegurar el contenedor ante aperturas accidentales o no autorizadas, existen candados en material plástico con y sin indicador químico.
- Las válvulas, que son barreras permanentes, requieren un mantenimiento programado que garantice su correcto funcionamiento.
- Envasado de textil (batas, paños, sábanas, gasas y compresas) se realiza con un sistema de barrera estéril (papel crepado o tejido sin tejer), además de un empaquetado de protección para su transporte y almacenaje.
- Empaquetado para esterilización por gas plasma
Solamente se pueden utilizar bolsas mixtas de papel tipo Tyvek®. Dicha bolsa pretende conseguir una mayor resistencia mecánica de la capa no transparente, al estar fabricada con una lámina de polietileno. En el interior de la bolsa siempre se introducirá un indicador químico de proceso.
- Empaquetado para esterilización por óxido de etileno
Se puede utilizar bolsa de papel mixto y bolsa de papel tipo Tyvek®. En el interior de la bolsa siempre se introducirá un indicador químico de proceso.
Actualmente las centrales de esterilización tienden a tener trazabilidad absoluta para saber piezas contenidas en cada paquete, ciclos realizados, por quien, donde va destinado el paquete, etc para tener un control total. Los paquetes se deben identificar con los siguientes datos: material introducido, fecha de envasado, fecha de esterilización, fecha de caducidad, número de esterilizador en el que se realizó, número de ciclo y tipo de programa, con los programas de trazabilidad también incluirán un código de barras que se podrá unir a la historia del paciente como las de los paquetes de cobertura.
En los paquetes y contenedores se debe colocar los indicadores de esterilización, se introducirá un control químico según el tipo de esterilización que el personal de quirófano deberá verificar siempre, antes de la utilización del material.
El sellado en el tejido sin tejer y en el polipropileno es mediante cinta adhesiva, el caso del tejido sin tejer la cinta lleva control químico que vira al esterilizarse, en el polipropileno no lo lleva y se debe poner una etiqueta para el control exterior. Las bolsas se sellan mediante termosellado. Los contenedores se cierran para evitar la apertura accidental con un precinto de apertura rápida.
Una vez realizados los paquetes se pasa a la zona de esterilización. Los métodos de esterilización pueden ser por:
- Calor que puede ser húmedo la autoclave, o seco la estufa de poupinel (en desuso en la actualidad) o túneles de esterilización.
- Esterilización química por óxido de etileno, peróxido de hidrogeno en estado gas plasma, ácido peracético y vapor de formaldehido (en desuso ya que primeramente la FDA lo dejo de considerar agente esterilizante y en Europa después).
- Radiación gamma, de electrones o ultravioleta.
Todos los métodos de esterilización deben tener controles del ciclo de esterilización. Si sucede un mal funcionamiento durante un ciclo, la carga debe ser considerada no estéril y los paquetes deben ser nuevamente empaquetados y reprocesados. Estos son de diferentes tipos:
CONTROL FÍSICO
Los controles físicos los realizan los propios equipos de esterilización y permiten visualizar si se cumplen los parámetros necesarios del proceso (temperatura, presión, humedad, tiempos).
En el registro del proceso se debe incluir la siguiente información: el número de identificación del esterilizador, la carga, la fecha, la hora y el nombre del operador de la central de esterilización que ha interpretado los registros obtenidos.
Los controles físicos son de gran utilidad, pero no son suficientes como “indicador de esterilización”, ya que pueden no reflejar lo que ocurre durante el proceso, a consecuencia de factores como son el tamaño de la carga o la presencia de materia orgánica no detectada.
CONTROL QUÍMICO
Los indicadores químicos son sustancias empleadas para controlar uno o más parámetros del proceso de esterilización, con el propósito de detectar fallos en el paquete, en la carga o en la función del esterilizador. Son de fácil lectura e interpretación y deben ser estables en el tiempo. Ningún indicador químico verifica que un dispositivo está realmente estéril.
Control químico externo
Se utiliza para identificar los artículos procesados con un simple vistazo y para tener la seguridad de que el paquete ha sido expuesto al proceso de esterilización, sin embargo, no establece si se han cumplido los parámetros para una esterilización adecuada del material de su interior.

Los indicadores químicos externos cambian de color cuando han sido expuestos al agente esterilizante. Estos indicadores son específicos para cada sistema de esterilización y van impresos en la cinta adhesiva que sella los paquetes, en las bolsas mixtas, en las etiquetas de identificación que se colocan en el exterior del contenedor o los paquetes e, incluso, en los propios candados de los contenedores.
Si los indicadores no han virado correctamente, o si existe duda, ese instrumental debe considerarse como no estéril, y se volverá a empaquetar y a esterilizar antes de su uso.
Se deben revisar al final del proceso de esterilización, antes de la distribución del material y antes de la utilización del material.
Control químico interno
Se utilizan para comprobar las variables críticas del proceso de esterilización, colocándolo en el punto de peor acceso del agente esterilizante en los paquetes, contenedores y/o bolsas.
Actualmente, se utilizan indicadores químicos multi-parámetros (temperatura, concentración y tiempo), distintos según el proceso de esterilización a que van a ser sometidos.
En las bolsas y paquetes, los indicadores químicos internos, son incluidos en la central de esterilización; pero los de los contenedores de instrumental quirúrgico se introducen al hacer la caja en el bloque quirúrgico.
Los indicadores integradores, que emulan a los indicadores biológicos, son los de elección para los contenedores de instrumental quirúrgico, aunque ya se recomienda incluirlos en todos los paquetes de más de 30 litros y en todos aquellos con material implantable.
Estos indicadores no pueden ser retirados sin alterar las condiciones de esterilidad, por lo que son comprobados en el momento de su uso.
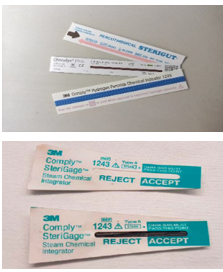
En el caso de no ser correctos, el contenido del paquete se considera no estéril y será devuelto a la central de esterilización junto con el indicador.
En la central se procederá a valorar el registro físico del ciclo de esterilización en el que se procesó dicho paquete, abrir varios paquetes de esa misma carga y examinar los controles químicos internos y si se observan varios fallos en la misma carga, retirar todos los artículos de esa carga.
CONTROL BIOLÓGICO
Los controles biológicos son los únicos que están diseñados para validar la eficacia de un proceso de esterilización. Son dispositivos preparados con esporas no patógenas y altamente resistentes a los procesos de esterilización. Las esporas utilizadas son de Bacillus atrophaeus para óxido de etileno y de Geobacillus stearothermophilus para vapor y para gas plasma.
Según las recomendaciones del grupo español de estudio sobre esterilización, se colocará un control biológico (espora) en cada autoclave diariamente y en cada ciclo a baja temperatura.
Los indicadores biológicos se ubicarán dentro de una bolsa mixta o dentro de una jeringuilla con la capacidad adecuada. En la etiqueta del control biológico se registrará el equipo, el número de ciclo y la fecha.
Los controles biológicos se procesan en la central de esterilización, en incubadoras específicas según el tipo de control biológico. El procedimiento es:
Se extrae el control del esterilizador, se activa la ampolla rompiéndola poniendo en contacto el medio de cultivo con las esporas, se agita el vial y se coloca en el pocillo de la incubadora. Se deja en la incubadora el tiempo indicado por el fabricante que es aproximadamente 3 horas para autoclaves, 4 horas para el óxido de etileno y 24 minutos para gas-plasma. Tras la incubación generan una luz verde si la espora no crece y una luz roja si se detecta crecimiento.
En caso de obtener un control biológico incorrecto:
- Comprobar el resultado del resto de controles de esa carga: Bowie-Dick, controles físicos, controles químicos externos e internos.
- Detener el funcionamiento del esterilizador y revisar.
- Realizar un ciclo en vacío y comprobar los parámetros físicos.
- Retirar la carga que todavía no se haya utilizado, para reempaquetarla y reesterilizarla.
- Si los artículos ya han sido usados en una intervención quirúrgica, notificar al servicio de Medicina Preventiva y al médico responsable para que el paciente sea vigilado o reciba tratamiento profiláctico.
PRUEBA DE BOWIE-DICK
No es una prueba del control de la esterilidad, sino del correcto funcionamiento de la etapa de extracción de aire de la cámara del autoclave y, consecuentemente, de la buena penetración del vapor en el paquete de prueba, en esta prueba se controla que después de la fase de pre vacío no existe aire en la cámara.
La prueba o control de Bowie-Dick es de obligado cumplimiento y de realización diaria (primer programa del día). Se deberá realizar también tras cualquier avería o reparación.
El paquete de prueba se debe colocar horizontalmente en la base del autoclave, en la parte frontal, cerca de la puerta y sobre el drenaje, en una cámara vacía y en un ciclo específico de 134C con un tiempo de exposición de 3-5 minutos.
- Prueba correcta: el indicador habrá virado hacia otra tonalidad de manera uniforme y en toda su extensión.
- Prueba incorrecta: se manifiesta por un color más tenue que el indicado por el fabricante o por la aparición de manchas o zonas de distinta densidad.
6.2 METODOS DE ESTERILIZACIÓN
A continuación, se desarrollarán los métodos que en algún momento se han utilizado con los productos sanitarios, sabiendo que los más utilizados actualmente son: autoclave, peróxido de hidrogeno en fase gas plasma, ácido peracetico y el óxido de etileno.
Calor húmedo. Autoclave
Una autoclave es un equipo que utiliza vapor de agua a alta presión y temperatura para destruir los microrganismos e inactivar las células por desnaturalización y coagulación de las proteínas consiguiendo la esterilización. Es el método más utilizado en los centros sanitarios.
Se utiliza de forma general en textiles, instrumental, vidrios, cerámica, prótesis e implantes, en general material que soporte las altas temperaturas sin deteriorase.
El autoclave está formado por:
- Dos puertas, esta sería la situación ideal donde una puerta es de entrada y otra de salida, aunque no siempre se tiene esta posibilidad.
- Cámara de acero inoxidable con separación de agua y diferentes estantes para poder situar el material en los contenedores.
- Recamara. Esta recubre la cámara y es donde se produce el vapor de agua por medio de un generador que debe garantizar la ausencia de puntos fríos.
- Filtros de aire y vapor.
- Pantalla para visualizar cómo va el ciclo con las diferentes alarmas: fase, temperatura de la cámara, tiempo restante, mensajes de error.
- Opcionalmente pueden tener conexión informática para poder mantener la trazabilidad.
Existen controles que se deben realizar a las autoclaves para comprobar su buen funcionamiento:
- Todos los días antes de su utilización se debe realizar la prueba de Bowie&Dick.
- En cada ciclo tendrá control químico donde se comprueba que el ciclo ha sido correcto por el cambio de color de la tira incluida.
- Control físico en cada ciclo con las gráficas del ciclo donde se verá la temperatura, presión, humedad y control de vapor en todas las fases del ciclo.
- Biológico se deben poner semanalmente y en cada ciclo en el caso de implantes.
Las fases de los ciclos del autoclave son acondicionamiento, meseta de esterilización, desvaporización y secado, siendo la fase del secado la que puede variar la duración total ya que el material debe salir totalmente seco y según la cantidad a esterilizar puede variar. El autoclave dispone de varios tipos de ciclos o programas con tiempo diferente según fabricante y la carga introducida, por ello se indican tiempos aproximados en cada ciclo. Los ciclos que siempre deben estar incluidos son:
- Estandar a 121° se utiliza para material que no soporta más temperatura según las indicaciones del fabricante como las gomas y el caucho. Tiene una duración aproximada de 1 hora.
- Estandar a 134° se utiliza para instrumental suelto o textil. Tiene una duración aproximada de 45 minutos.
- Contenedores a 134° Tiene una duración aproximada de 1 hora y 15 minutos. Según los contenedores el tiempo del secado se puede tener que alargar.
- Rápido a 134°. Se utiliza para urgencias envasado en bolsa. El material no se puede almacenar, se tiene que utilizar en las 2 horas siguientes a terminar la esterilización, en el caso de no ser utilizado se debe volver a esterilizar con un ciclo completo. Tiene una duración aproximada de 20 minutos, el tiempo es muy corto por no tener fase de secado y por eso no se puede almacenar.
- Especiales priones a 134°. Tiene una duración aproximada de 1 hora y 30 minutos
Las ventajas de este procedimiento son que es barato, rápido, eficiente, no es toxico y es fácil de monitorizar. Las desventajas que presenta son no se puede utilizar con material termosensible, ni en polvos, ni sustancias oleosas o grasas y no se puede utilizar en instrumental cromado.
Calor seco
Se inactivan las células mediante oxidación por cambio de calor de 160° a 280° durante un tiempo entre 30 minutos y 2 horas. Según la temperatura que se utilice se deberá tener más o menos tiempo determinando así los ciclos. Se puede realizar mediante:
- Horno o estufa de calor. Se debe colocar correctamente los materiales para que el flujo de aire circule correctamente. Proceso discontinuo con temperatura de 120-180°. Poupinel.
- Túnel de esterilización. Se usa a nivel industrial es un proceso continuo con temperatura superior a 250° pudiendo alcanzar los 300°.
Este método es cada vez menos utilizado, casi en desuso. Se utiliza de forma general en materiales resistentes al calor como vaselinas, sustancias grasas u oleosas, talco, instrumental cromado o niquelado, envases de vidrio, cerámico o pyrex.
Los controles que se deben realizar para comprobar su buen funcionamiento:
- Control físico en cada ciclo con sensores de temperatura y tiempo para determinar la homogenidad en todas las zonas durante el proceso.
- Biológico en cada ciclo.
- Sistemas de filtración con controles de ausencia de partículas e integridad de los filtros.
Las ventajas de este procedimiento son que es barato, no es toxico, no es corrosivo y puede alcanzar instrumentos que no pueden desarmarse. Las desventajas que presenta son es un proceso más lento, está limitado a materiales empaquetados y de unos tipos concretos.
Óxido de etileno
Este método inactivo por medio químico, óxido de etileno gas, que afecta a la capacidad metabólica y la reproducción celular. El óxido de etileno se presenta licuado a presión en diferentes formatos capsulas o tanques a nivel industrial y necesita una instalación exclusiva con buena ventilación y sistema de extracción diferente a los de otros sistemas del centro. Trabaja a bajas temperaturas 30-60° con un tiempo largo entre 2 y 5 horas y es obligatoria aireación posterior de mínimo 12 horas. Esta cada vez más en desuso a nivel hospitalario y solo se permite utilizar para material que no pueda ser esterilizado de otra forma por que pueda deteriorarse.
Los controles que se deben realizar para comprobar su buen funcionamiento:
- Control físico en cada ciclo con sensores de temperatura, presión, tiempo y humedad.
- En cada ciclo tendrá control químico con el cambio de color de la tira incluida.
- Biológico en cada ciclo.
Las ventajas de este procedimiento son que se puede utilizar con casi todos los materiales, permite esterilizar material con lumen por su buena penetración y tiene un amplio margen de seguridad de esterilización. Las desventajas que presenta son que es un proceso caro, toxico para los humanos, más lento, requiere aireación, altamente inflamable y explosivo lo que conlleva control de los niveles de gas y tiene riesgo de toxicidad ambiental.
Peróxido de hidrogeno en estado gas plasma
Es una esterilización a baja temperatura por medio del peróxido de hidrogeno en fase gas plasma, es un estado entre líquido y gas, que se presenta en cartuchos. Se realiza una vaporización del peróxido a baja temperatura, no más de 60° y baja presión.
En general si un material se puede esterilizar con óxido de etileno, se puede realizar con este método con la ventaja que no requiere aireación. Se puede utilizar para material con lumen de acero, polietileno y teflón y material que no pueda ser esterilizado en autoclave. Según el material a esterilizar se programará el ciclo que variará sobre todo el tiempo del mismo de 30 a 60 minutos. El ciclo consta de tres fases consiguiendo estado gas plasma en dos de ellas.
Los controles que se deben realizar para comprobar su buen funcionamiento:
- Control físico en cada ciclo con sensores de temperatura, presión y tiempo.
- Indicador químico.
- Biológico en cada ciclo.
Las ventajas de este procedimiento son que se puede utilizar con casi todos los materiales, permite esterilizar material con lumen por su buena penetración, los ciclos son cortos y tiene un amplio margen de seguridad de esterilización. Las desventajas que presenta son que es caro, en lúmenes estrechos necesita adaptador, no funciona con humedad se inactiva y se necesita empaquetado especial tipo Tyvek que no contenga celulosa ya que se inactiva con materiales altamente absorbentes.
Ácido peracético
Es una esterilización a baja temperatura en punto de uso que se realiza por inmersión en cubeta con ácido peracético a baja temperatura, entre 50-55°, para material suelto que no necesita ser secado previamente. Se utiliza sobre todo para material termosensible. El tiempo que debe estar sumergido el material es de 30 minutos.
El proceso es estandarizado y se debe controlar la temperatura y el tiempo de exposición.
Las ventajas son que es muy rápido, sirve para todo material que se pueda sumergir, se realiza en punto de uso a baja temperatura, no deja residuo, es efectivo con restos de materia orgánica y no daña el medio ambiente evitando efectos adversos al operador. Las desventajas son que no tiene una monitorización precisa del proceso, solo se puede usar en material sumergible, es corrosivo, en cada ciclo entra poco material, produce un fuerte olor y es caro.
Vapor de formaldehido
En este método se utiliza solución de formaldehido al 2-3% en estado vapor con vapor de agua a baja temperatura (50-60°). Se puede utilizar en materiales termolábiles, aunque está cada vez más en desuso ya que primeramente la FDA lo dejo de considerar agente esterilizante y en Europa después, en España no está homologado.
Los controles que se deben tener son registro continuo de temperatura, presión y tiempo, indicadores físicos, químicos y biológicos.
Las ventajas no son muchas para los inconvenientes que presenta y por ello se retiró. Ventajas son compatibilidad con muchos materiales y procesos cortos con fácil instalación y manejo. Los inconvenientes son complejidad de control de las variables, necesita control ambiental, inflamable, toxico e irritante y necesita largo tiempo de aireación.
Radiación ionizante
El sistema produce la ionización del ADN de los microorganismos que produce una rotura de las cadenas y formación de enlaces transversales impidiendo la multiplicación celular. Los más sensibles son los gram negativos, seguidos de los hongos, levaduras, virus y formas esporuladas. Se puede utilizar en material plástico, prótesis, material de desechable, evitado material metálico porque lo calienta. No está considerado, por la F D A y cada vez más en entredicho por la CEE, como agente esterilizante.
Se debe controlar la dosis de radiación, tiempo y el poder de penetración además de controles químicos y biológicos con tiras de bacilluspumillus.
Las ventajas que presenta este método son que es ecológico, no toxico y fácil de controlar. Las desventajas son que necesita instalaciones especiales y operadores especializados y provoca calor en el material metálico.
6.3 REPROCESAMIENTO DE MATERIAL. PRODUCTOS SANIATRIOS IN HOUSE
Normativa
El Reglamento (UE) 2017/745 armoniza las normas aplicables a la introducción en el mercado y la puesta en servicio en la Unión Europea de productos sanitarios y sus accesorios, permitiendo así que los mismos se acojan al principio de libre circulación de mercancías, consolida el criterio normativo aplicable a una serie de materias relevantes, como son la supervisión de los organismos notificados, los procedimientos de evaluación de la conformidad, las investigaciones clínicas y evaluación clínica, la vigilancia y el control del mercado e introduce disposiciones que garantizan la transparencia y trazabilidad de los productos sanitarios, a través de la base de datos europea Eudamed, el sistema de identificación única (Sistema UDI) siendo todo ello de aplicación directa a los estados miembros pero se hace preciso regular a nivel nacional los aspectos que la norma europea deja a la regulación de cada Estado miembro. Por ello, en España se aprueba el Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios. con la finalidad de regular los procedimientos para la fabricación de productos para su uso en el propio centro sanitario y para el reprocesamiento de productos de un solo uso y su utilización.
El RD recoge las condiciones necesarias para la fabricación, agrupación, esterilización, e importación de productos sanitarios que deben reunir las empresas que se dediquen a estas actividades, así como al procedimiento a seguir para el otorgamiento de la licencia previa de funcionamiento y destaca, como novedad, la regulación del reprocesamiento de productos sanitarios de un solo uso en hospitales, sólo se autoriza con los requisitos previstos de este RD e igualmente establece las condiciones de utilización de los productos reprocesados. Los sujetos autorizados a realizar estas actividades de reprocesamiento son los fabricantes y los hospitales, si bien, en cuanto a estos últimos, su actividad en este ámbito precisa de una mayor concreción técnica que se especifica será regulada por el Ministerio de Sanidad.
También recoge la posibilidad de la fabricación por los hospitales de productos para su propio y exclusivo uso, establece la obligación de comunicar el inicio de la actividad. Este tipo de fabricación y utilización exclusiva en hospitales no está dirigida a una posterior comercialización ni beneficio económico y redunda en el interés directo de un grupo específico de pacientes para los que no existen alternativas en el mercado, por lo que requiere de la correspondiente agilidad y flexibilidad, en el interés de los pacientes, siempre garantizando el adecuado funcionamiento y seguridad de los productos.
La AEMPS en noviembre de 2024 ha puesto en marcha la aplicación informática para poder comunicar la actividad de fabricación in house de productos sanitarios por parte de los hospitales, cada hospital deberá de designar a un coordinador dentro de su hospital para solicitar y gestionar el alta en la aplicación. Esto se podrá realizar teniendo en cuenta lo recogido en el Reglamento ya que indica que es importante resaltar que esta fabricación debe tener como finalidad satisfacer las necesidades específicas del grupo de pacientes al que se destinan los productos, las cuales no pueden satisfacerse o no pueden satisfacerse con el nivel de funcionamiento adecuado, mediante otro producto con marcado CE comercializado. De acuerdo al Real Decreto, esta actividad solo puede ser realizada por hospitales y no se permite fabricar productos de las clases IIb, III, ni productos implantables y tampoco aplica a productos sanitarios a medida, los cuales requieren de licencia previa de funcionamiento para su fabricación por parte de la Comunidad Autónoma.
Respeto al reprocesamiento y fabricación in house se deben tener en cuenta algunas definiciones del Reglamento y la AEMPS como son:
- Reprocesamiento: un proceso llevado a cabo con un producto usado para hacer posible su reutilización segura, incluida la limpieza, desinfección, esterilización y procedimientos asociados, así como los ensayos y la restauración de la seguridad técnica y funcional del producto usado.
- Producto in house: producto fabricado y utilizado exclusivamente en un mismo centro sanitario establecido en la Unión Europea y que cumple las condiciones dispuestas en el Artículo 5(5) del Reglamento (UE) 2017/745.
- Producto de un solo uso: el destinado a usarse en una única persona durante un procedimiento único.
- Deficiencia de un producto: toda inadecuación de la identidad, la calidad, la durabilidad, la fiabilidad, la seguridad o la eficacia de un producto en investigación, con inclusión de su funcionamiento defectuoso, errores de utilización o la inadecuación de la información facilitada por el fabricante.
- Incidente: todo funcionamiento defectuoso o deterioro de las características o el funcionamiento de un producto comercializado, incluidos los errores de uso debidos a características ergonómicas, así como cualquier inadecuación de la información facilitada por el fabricante o cualquier efecto colateral indeseable.
- Incidente grave: todo incidente que, directa o indirectamente, haya podido tener o haya tenido alguna de las siguientes consecuencias: a) el fallecimiento de un paciente, usuario u otra persona, b) el deterioro grave, temporal o permanente, de la salud de un paciente, usuario u otra persona, c) una grave amenaza para la salud pública.
- Acontecimiento adverso: todo incidente médico perjudicial, enfermedad o lesión no prevista o signo clínico desfavorable, incluido un resultado analítico anómalo, que se produce en sujetos, usuarios u otras personas en el contexto de una investigación clínica, tenga o no relación con el producto en investigación.
- Acontecimiento adverso grave: todo acontecimiento adverso que ha tenido alguna de las siguientes consecuencias: a) fallecimiento, b) deterioro grave de la salud del sujeto que cause: i) enfermedad o lesión potencialmente mortales, ii) deterioro permanente de una función corporal o de una estructura corporal, iii) hospitalización o prolongación de la hospitalización del paciente, iv) intervención médica o quirúrgica para evitar una enfermedad o lesión potencialmente mortales o el deterioro permanente de una función corporal o de una estructura corporal, v) enfermedad crónica, c) sufrimiento fetal, muerte fetal o una deficiencia física o psíquica o malformación congénitas.
El reprocesamiento de un producto conlleva una serie de procesos y debe conseguir la misma calidad que el nuevo y con los mismos controles, por ello el reprocesador asume el papel de fabricante y la responsabilidad recae en la persona que decide realizarlo. En los hospitales realizamos reprocesamiento de materiales reutilizables, pero no estamos autorizados para materiales de un solo uso.
Los principales problemas que se pueden encontrar son:
- Deterioro del PS por que no se consiga mantener sus características iniciales como estructura, corte, corrosión del material…Existen algunos materiales que el fabrícate establece, así figura en su documentación, un número máximo de reprocesamientos y como debe realizarse.
- Riesgo de infección por una mala esterilización en material no considerado para ello.
Sabiendo lo que se entiende por reprocesamiento debemos concluir que los materiales de un solo uso, solo se pueden volver a procesar si eres fabricante ya que los PS reprocesados deben tener las mismas características y calidad que el nuevo sin ser un riesgo para el paciente. En los hospitales realizamos reprocesamiento de materiales reutilizables, pero no estamos autorizados para materiales de un solo uso porque actualmente somos reprocesadores, no fabricantes.
La fabricación in house según indica la AEMPS, puede incluir:
“- Fabricar un producto partiendo de materias primas, de partes o componentes de un producto o de otro tipo de productos diferentes de los productos sanitarios y los productos del Anexo XVI, o partiendo de un producto existente o de otro tipo de productos existentes diferentes de los productos sanitarios y los productos del Anexo XVI.
Combinar un producto con otro producto o con otro tipo de productos diferentes de los productos sanitarios y los productos del Anexo XVI, cuando la combinación crea un nuevo producto.
Modificar un producto existente para crear uno nuevo.
Los productos deben fabricarse y utilizarse dentro del mismo hospital. El uso debe entenderse como un uso físico ó remoto, en el caso de software, siempre que no se pongan a disposición de otras entidades legales”
Los hospitales deben notificar a la AEMPS la comunicación de querer realizar la actividad para conseguir el permiso para la misma. Se cumplimentará aportando los siguientes datos:
- Persona responsable
- Declaración pública respecto a la fabricación y uso de los productos in house,
- Justificación de no existir alternativas en el mercado,
- Descripción sobre el uso de los productos que incluya la justificación de la fabricación y uso,
- Declaración de la disponibilidad de documentación del producto a fabricar, de las instalaciones de fabricación, proceso
- Procedimiento del seguimiento de la experiencia adquirida con el uso del producto.
Cualquier modificación en la fabricación debe ser reportada a la AEMPS también en el caso de cesar la actividad.
La fabricación in house abre una nueva posibilidad, pero lo importante es que quien fabrique debe aportar las garantías sanitarias oportunas al igual que lo realizan las fábricas de material sanitario.
BIBLIOGRAFÍA
- https://es.linkedin.com/pulse/el-instrumental-quir%C3%BArgico-algo-de-su-historia-albarracin-miranda.
- historia.nationalgeographic.com.es/a/sofisticacion-antiguo-egipto-protesis-hace-3000-anos_11639
- portalcomunicacion.uah.es/diario-digital/actualidad/medicina-medieval-una-etapa-oscura-pero-llena-de-supersticiones/
- www.salusplay.com/apuntes/quirofano-y-anestesia/tema-3-instrumentacion
- wfsahq.org/es/about/history/history-of-anaesthesia
- www.bbvaopenmind.com/ciencia/investigacion/joseph-lister-el-hombre-que-esterilizo-la-cirugia/
- www.revistamedicinaycultura.fmposgrado.unam.mx/index.php/2023/01/24/joseph-lister-biografia/
- identyd.com/historia-autoclave/
- www.elsevier.es/es-revista-revista-argentina-radiologia-383-articulo-wilhelm-conrad-roentgen-el-descubrimiento-S0048761916301545
- www.fda.gov/medical-devices/overview-device-regulation/history-medical-device-regulation-oversight-united-states
- formasefh.sefh.es/tecnifarmh/curso-productos-sanitarios/curso-productos-sanitarios.pdf
- www.aemps.gob.es/la-aemps/legislacion/legislacion-sobre-productos-sanitarios/
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- www.aemps.gob.es/productos-sanitarios/investigacionclinica-productossanitarios/
- UNE-EN ISO 14971:2020 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN 62366-1:2015/A1:2020 (Ratificada) Productos sanitarios. Parte 1: Aplicación de la ingeniería de usabilidad a los productos sanitarios.
- Aplicación de la gestión de riesgos a los productos sanitarios (une.org)
- 231219_GuiaValidacion_Tec_Sanitarias-1.pdf (inndromeda.es)
- Ciclo de vida de producto sanitario archivos - Consultoría de producto sanitario
- Ciclo de vida de producto sanitario y gestión de riesgos - Fernando Atienza
- Producto sanitario definición, características y usos (productossanitarios.net)
- Risk Management in the Medical Device Industry (dqsglobal.com)
- Design Thinking en Producto Sanitario ¿Qué es idear, prototipar y testar? (ambit-bst.com)
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Ley 29/2006, de 26 de julio de garantías y uso racional del medicamento.
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS): https://www.aemps.gob.es/
- Federación Española de Empresas de Tecnología Sanitaria (FENIN): https://www.fenin.es/
- Ministerio de Ciencia e Innovación (PERTE Salud de Vanguardia): https://www.ciencia.gob.es/
- European Commission. (2021). Guidance on the application of the MDR and IVDR during the COVID-19 pandemic. Publications Office of the European Union. https://health.ec.europa.eu/system/files/2021-05/md_guidance_md_011_en_0.pdf
- Ghisellini, P., Cialani, C., & Ulgiati, S. (2016). A review on circular economy: The expected transition to a balanced interplay of environmental and economic systems. Journal of Cleaner Production, 114, 11-32. https://doi.org/10.1016/j.jclepro.2015.09.007
- Organización Internacional del Trabajo (OIT). (2023). Declaración de principios y derechos fundamentales en el trabajo. https://www.ilo.org/global/lang--es/index.htm
- Organización Mundial de la Salud (OMS). (2022). Health product policy and standards. https://www.who.int/teams/health-product-and-policy-standards
- Parlamento Europeo y Consejo de la Unión Europea. (2017). Reglamento (UE) 2017/745 del Parlamento Europeo y del Consejo de 5 de abril de 2017 sobre los productos sanitarios. Diario Oficial de la Unión Europea. https://eur-lex.europa.eu/legal-content/ES/TXT/?uri=CELEX%3A32017R0745
- Porter, M. E., & Kramer, M. R. (2011). Creating shared value. Harvard Business Review, 89(1/2), 62-77.
- OMS. (2015). Ética de la salud mundial: cuestiones clave.
- Declaración de Helsinki de la AMM – Principios éticos para las investigaciones médicas con participantes humanos – WMA – The World Medical Association.
- Global health ethics: key issues (who.int)
- https://lac.saludsindanio.org/cambio-climatico-y-salud/huella-climatica-del-sector-salud
- https://www.miteco.gob.es/content/dam/miteco/es/cambio-climatico/temas/mitigacion-politicas-y-medidas/docuementoexplicativosello_tcm30-479000.pdf
- https://accionclimaticaensalud.org/sites/default/files/2021-06/huellaclimatica.pdf
- Estrategia de Economía Circular de Euskadi 2030 - Prevención de la contaminación, inspección y control ambiental - Euskadi.eus
- Residuos Sanitarios (miteco.gob.es)
- Guía de procedimientos de esterilización en el medio hospitalario. file:///C:/Users/50820026P/Documents/OneDrive%20-%20Madrid%20Digital/PERSONAL/MASTER%20ordenador%20sollube/2.%20TEMARIO%20MASTER%20FARMA%20Y%20PS/procedementos_esterilizacion.pdf
- Curso básico de Gestion de Productos Sanitarios ANECORM
- https://www.aemps.gob.es/informa/la-aemps-pone-en-marcha-una-nueva-aplicacion-para-la-comunicacion-de-fabricacion-de-productos-sanitarios-in-house-por-hospitales/
- REGLAMENTO (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) n.o 178/ 2002 y el Reglamento (CE) n.o 1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo (boe.es)
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- Reglamento (UE) 2017/ 745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO - de 5 de abril de 2017 - sobre los productos sanitarios, por el que se modifican la Directiva 2001/ 83/ CE, el Reglamento (CE) nº178/ 2002 y el Reglamento (CE) nº1223/ 2009 y por el que se derogan las Directivas 90/ 385/ CEE y 93/ 42/ CEE del Consejo
- Reglamento (UE) 2017/746 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios para diagnóstico in vitro y por el que se derogan la Directiva 98/79/CE y la Decisión 2010/227/UE de la Comisión.
- Reglamento (UE) 2022/112 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 25 de enero de 2022.
- Reglamento (UE) 2023/607 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 15 de marzo de 2023.
- Reglamento (UE) 2024/1860 DEL PARLAMENTO EUROPEO Y DEL CONSEJO, de 13 de junio de 2024.
- Real Decreto 192/2023, de 21 de marzo, por el que se regulan los productos sanitarios.
- UNE-EN ISO 9001:2015. Sistemas de gestión de la calidad. Requisitos (ISO 9001:2015)
- UNE-EN ISO 13485:2018 Productos sanitarios. Sistemas de gestión de la calidad. Requisitos para fines reglamentarios
- UNE-EN ISO 14971:2019 Dispositivos médicos/productos sanitarios (MD). Aplicación de la gestión de riesgos a los MD.
- UNE-EN ISO 10993-1:2021 Evaluación biológica de productos sanitarios.
- Directiva 2010/32/UE Protección frente a lesiones por objetos cortopunzantes en el sector sanitario
- Real Decreto 664/1997 modificado por RD 598/2015 Obligación de adoptar medidas técnicas y
- Organizativas específicas para prevenir pinchazos accidentales