1. DEFINICIÓN DE PRODUCTO SANITARIO
Los productos sanitarios son los materiales utilizados en un centro sanitario (hospitales, atención primaria, centros de crónicos) y servicios externos de urgencias. Son utilizados para prevención, diagnóstico, tratamiento y control de pacientes.
A nivel europeo, desde 1990, el marco normativo europeo para productos sanitarios se recoge en las siguientes Directivas y Reglamentos, con sus modificaciones posteriores:
- Directiva 90/42/CEE de Productos sanitarios
- Directiva 90/385/CEE de Productos Sanitarios implantes activos
- Directiva 98/79/CEE de productos sanitarios para diagnostico in vitro.
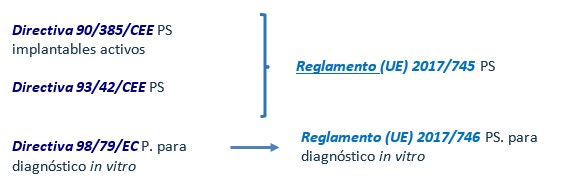
Actualmente existe un nuevo REGLAMENTO (UE) 2017/745 DEL PARLAMENTO EUROPEO Y DEL CONSEJO de 5 de abril de 2017 sobre los productos sanitarios, tendría que haber entrado en vigor el 26 de mayo de 2020 pero por la pandemia entrara en vigor el 26 de mayo 2021, este reglamento unifica las Directivas 90/42/CEE y 90/385/CEE y actualiza los puntos existentes en las anteriores Directivas, sobretodo en la comercialización de los productos y en la función de los organismos reguladores e incluye alguno nuevo como es la identificación y trazabilidad de los productos sanitarios mediante el sistema UDI.
Los productos sanitarios para diagnostico in vitro también tiene una nueva legislación Reglamento (UE) 2017/746 Productos sanitarios para diagnostico in vitro. Esta entrará en vigor el 26 de mayo de 2022.
En España, se desarrollaron diversos Reales Decretos, partiendo de las Directivas, siendo los actuales;
- RD 1591/2009 de 6 de noviembre de 2009, por el que se regulan los productos sanitarios
- RD 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos.
- Real Decreto 1193/2012, de 3 de agosto, por el que se modifica el Real Decreto 1662/2000, de 29 de septiembre, sobre productos sanitarios para diagnostico “in vitro”
A día de hoy se están desarrollando nuevos RD, aunque no se sabe con certeza cuando verán la luz, ante esta situación solo podemos afirmar que gran parte del Reglamento Europeo entrara en vigor en mayo de 2021 y las empresas tendrán que estar adoptadas para su cumplimiento ya que los Reglamentos son actos jurídicos que se aplican de manera automática y uniforme en todos los países de la Unión Europea desde su entrada en vigor, sin necesidad de incorporación al derecho nacional.
Los nuevos Reglamentos definen los productos sanitarios, los productos sanitarios in vitro y los productos implantables;
“Producto Sanitario es cualquier instrumento, dispositivo, equipo, programa informático, implante, reactivo, material u otro artículo, destinado por el fabricante a ser utilizado en personas, por separado o en combinación, con alguno de los siguientes fines médicos específicos:
- Diagnóstico, prevención, seguimiento, predicción, pronóstico, tratamiento o alivio de una enfermedad.
- Diagnóstico, seguimiento, tratamiento, alivio o compensación de una lesión o de una discapacidad.
- Investigación, sustitución o modificación de la anatomía o de un proceso o estado fisiológico o patológico,
- Obtención de información mediante el examen in vitro de muestras procedentes del cuerpo humano, incluyendo donaciones de órganos, sangre y tejidos.
y que no ejerza la acción principal prevista en el interior o en la superficie del cuerpo humano por medios farmacológicos, inmunológicos ni metabólicos, pero a cuya función puedan contribuir tales mecanismos.”
También se consideran productos sanitarios:
- Los productos de control o apoyo a la concepción
- Los destinados específicamente a la limpieza, desinfección o esterilización de otros productos sanitarios.”
“Producto implantable: es todo producto, incluidos los que son absorbidos parcial o totalmente, que se destina a:
- ser introducido totalmente en el cuerpo humano o
- sustituir una superficie epitelial o la superficie ocular,
mediante intervención médica, y a permanecer en su lugar después de la intervención.
Se considerará asimismo producto implantable todo producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días; P impllantable activo”
“Producto sanitario para diagnóstico in vitro: cualquier producto sanitario que consista en un reactivo, producto reactivo, calibrador, material de control, kit, instrumento, aparato, pieza de equipo, programa informático o sistema, utilizado solo o en combinación, destinado por el fabricante a ser utilizado in vitro para el estudio de muestras procedentes del cuerpo humano, incluidas las donaciones de sangre y tejidos, única o principalmente con el fin de proporcionar información sobre uno o varios de los elementos siguientes:
a) relativa a un proceso o estado fisiológico o patológico;
b) relativa a deficiencias físicas o mentales congénitas;
c) relativa a la predisposición a una dolencia o enfermedad;
d) para determinar la seguridad y compatibilidad con posibles receptores;
e) para predecir la respuesta o reacción al tratamiento;
f) para establecer o supervisar las medidas terapéuticas.
Los recipientes para muestras se considerarán también productos sanitarios para diagnóstico in vitro”
2. CLASIFICACIÓNES DE PRODUCTOS UTILIZADOS EN LOS CENTROS SANITARIOS
La clasificación del material utilizado en los centros se debería considerar de dos formas diferentes, por la utilización en los centros (incluimos todo el material utilizado como por ejemplo también papelería) y por lo recogido los Reglamentos para producto sanitario.
2.1. CLASIFICACIÓN SEGÚN LA UTILIZACIÓN EN LOS CENTROS
Esta clasificación nos ayuda a entender la forma de funcionamiento de compras, almacén y reposición de las unidades, por ello se incluye material que se utiliza en los centros que no son productos sanitarios como tal, como por ejemplo la papelería.
Material fungible
Aquel que se consume durante su utilización o que debe ser desechado tras su uso, independientemente de su precio (actualmente en quirófanos, hemodinamia, radiología vascular se utilizan productos fungibles de alto valor económico). Por ello, quedan excluidos del inventario general de bienes y derechos.
Ejemplos; además del material de quirófanos, hemodinámica y radiología vascular, se incluye material de uso habitual en las unidades como gasas, sistemas de infusión, jeringas, etc.
Este es un material de uso corriente en todo centro sanitario y su adquisición se realiza a lo largo de todo el año.
Material inventariable
Aquel que va a permanecer en la entidad que lo adquiere y que no va dirigido a su venta. Tiene un periodo largo de uso, utilizandose en muchas ocasiones y generalmente tiene un coste elevado y necesita una valoración más profunda para su adquisición. Dentro de este grupo están el material clínico, material quirúrgico, mobiliario clínico.
Algunos ejemplos son: TAC, electrocardiógrafo, desfibrilador, pie de goteo, cama de paciente, mesilla de noche, mesa quirúrgica.
Según el material de que se trate tendrá una vida útil diferente, este periodo debe ser tal que asegure una buena amortización del producto. Su adquisición se realiza en el plan de inversiones anual o en planes específicos de la comunidad autónoma.
Pequeño material
Es material de oficina, repuestos, material clínico y de laboratorio de corta duración. Todo aquel que no permanece más de un ejercicio económico, así como la papelería, libros y revistas (estos últimos se inventarían en el catálogo de biblioteca y se asigna su adquisición a un ejercicio económico). Se usan en muchas ocasiones, el precio es bajo y se adquiere en conjunto o por separado. Es un grupo que se situaría en medio de los dos grandes grupos descritos. Se refiere a material no inventariable pero que puede ser reutilizado muchas veces. Su adquisición, normalmente, se realiza en el plan de inversiones anual
Por ejemplo; aparatos de tensión arterial, pinzas de curas, sujeciones mecánicas.
2.2. LA CLASIFICACIÓN SEGÚN LA NORMATIVA REGLAMENTO (UE) 2017/745.
Los productos se clasificarán en las clases I, IIa, IIb y III, teniendo en cuenta la finalidad prevista de los productos, sus riesgos inherentes, el tiempo de uso y la forma de uso. La clasificación se llevará a cabo de conformidad con el anexo VIII donde se desarrollan los diferentes criterios de clasificación y para ello debemos conocer ciertas definiciones y normas, en relación a las reglas de clasificación.
En caso de controversia entre el fabricante y el organismo notificado por razón de la aplicación de los criterios de clasificación, se remitirá para decisión a las autoridades competentes de las que dependa dicho organismo. En caso de tratarse de un organismo notificado español, se acudirá a la Agencia Española de Medicamentos y Productos Sanitarios.
La Agencia Española de Medicamentos y Productos Sanitarios decidirá sobre la clasificación que corresponde a los productos aplicando los criterios establecidos y podrá presentar a la Comisión Europea una solicitud cuando las reglas de clasificación requieran de una decisión para un determinado producto o para una excepción
DEFINICIONES
Por su duración
- Uso pasajero: destinados a utilizarse de forma continua durante menos de 60 minutos.
- Corto plazo: destinados normalmente a un uso continuado desde 60 minutos hasta 30 días.
- Uso prolongado: destinados normalmente a un uso continuado de más de 30 días.
Por su forma de uso
- Producto no invasivo.
- Producto invasivo: producto que penetra completa o parcialmente en el interior del cuerpo, bien por un orificio corporal o a través de la superficie del cuerpo. Por ejemplo, un catéter o una sonda nasogástrica
- Producto invasivo de tipo quirúrgico: Producto invasivo que penetra en el interior del cuerpo a través de la superficie corporal por medio de una intervención quirúrgica o en el contexto de una intervención quirúrgica.
- Producto implantable: producto, incluidos los que son absorbidos parcial o totalmente, que se destina a ser introducido totalmente en el cuerpo humano, o para sustituir una superficie epitelial o la superficie ocular, mediante intervención médica y a permanecer en su lugar después de la intervención. Se considerará asimismo producto implantable cualquier producto destinado a ser introducido parcialmente en el cuerpo humano mediante intervención médica y a permanecer en su lugar después de dicha intervención durante un período de al menos treinta días.
Otras definiciones:
- Instrumento quirúrgico reutilizable: instrumento destinado a fines quirúrgicos para cortar, perforar, serrar, escarificar, raspar, pinzar, retraer, recortar o procedimientos similares
- sin estar conectado a un producto sanitario activo, y destinado por el fabricante a ser reutilizado una vez efectuados los procedimientos adecuados tales como limpieza, desinfección y esterilización.
- Producto sanitario activo: humano a este efecto o por la gravedad, y que actúa cambiando la densidad de esta energía o convirtiendo esta energía. No se considerarán productos activos los productos destinados a transmitir energía, sustancias u otros elementos entre un producto activo y el paciente, sin ningún cambio significativo. Un programa informático también se considerará un producto activo;
- Producto activo terapéutico: cualquier producto activo, utilizado solo o en combinación con otros productos, destinado a sostener, modificar, sustituir o restaurar funciones o estructuras biológicas en el contexto del tratamiento o alivio de una enfermedad, lesión o deficiencia.
- Producto activo para diagnóstico y vigilancia: cualquier producto activo, utilizado solo o en combinación con otros productos, destinado a proporcionar información para la detección, el diagnóstico, la observación o el tratamiento de estados fisiológicos, de estados de salud, de enfermedades o de malformaciones congénitas
- Sistema circulatorio central: En el marco del Reglamento se entenderá los vasos sanguíneos siguientes: arteriae pulmonales, aorta ascendens, arcus aortae, aorta descendens hasta la bifurcatio aortae, arteriae coronariae, arteria carotis communis, arteria carotis externa, arteria carotis interna, arteriae cerebrales, truncus brachiocephalicus, venae cordis, venae pulmonales, vena cava superior y vena cava inferior.
- Sistema nervioso central: En el marco del Reglamento se entenderá “sistema nervioso central”, el cerebro, las meninges y la médula espinal.
NORMAS DE DESARROLLO
La aplicación de las reglas de clasificación se regirá por la finalidad prevista de los productos.
Si un producto en cuestión se destina a utilizarse en combinación con otro producto, las reglas de clasificación se aplicarán a cada uno de los productos por separado. Los accesorios para un producto sanitario y para un producto enumerado en el anexo XVI serán clasificados por sí mismos por separado del producto con el que se utilicen.
Los programas informáticos que sirvan para manejar un producto o tengan influencia en su utilización se incluirán en la misma clase que el producto. Si el programa informático es independiente de cualquier otro producto, será clasificado por sí mismo.
Si un producto no se destina a utilizarse exclusiva o principalmente en una parte específica del cuerpo, se considerará para la clasificación su utilización especificada más crítica.
Si para el mismo producto son aplicables varias reglas o si, dentro de la misma regla, son aplicables varias subreglas, teniendo en cuenta la finalidad prevista del producto, se aplicarán la regla y subregla más estricta que dé lugar a la clasificación más elevada.
Por utilización continua se entenderá:
a) toda la duración de uso del mismo producto, sin importar la interrupción temporal de su uso durante un procedimiento o la retirada temporal con fines de limpieza o desinfección del producto. El carácter temporal de la interrupción del uso o la retirada se establecerá en relación con la duración del uso antes y después del período en el que el uso se interrumpe o el producto se retira, y
b) el uso acumulado de un producto destinado por el fabricante a ser sustituido inmediatamente por otro del mismo tipo. 3.7. Se considerará que un producto permite un diagnóstico directo cuando proporciona el diagnóstico de la enfermedad o la afección en cuestión por sí mismo o cuando proporciona información decisiva para el diagnóstico.
Se considerará que un producto permite un diagnóstico directo cuando proporciona el diagnóstico de la enfermedad o la afección en cuestión por sí mismo o cuando proporciona información decisiva para el diagnóstico.
REGLAS DE CLASIFICACIÓN
Las reglas de clasificación están recogidas en el Reglamento en anexo VIII, capitulo III. Se desarrollan clasificadas en productos no invasivos, invasivos, activos y reglas especiales.
Una vez conocidas las definiciones, normas y reglas de clasificación, podemos saber que según los riesgos potenciales que pueden derivarse de la utilización de los productos sanitarios y aplicando las reglas de decisión que se basan en la vulnerabilidad del cuerpo humano, los productos sanitarios se clasifican siguiendo lo que indica el Reglamento Reglamento (UE) 2017/745
Se puede indicar a modo orientativo y no exhaustivo debido a la complejidad de los criterios y reglas de clasificación que se encuentran en los textos, la siguiente clasificación:
En general los productos de clase I son productos que corresponden a procedimientos con menor riesgo. Según aumenta la clasificación aumenta el riesgo, por ello, los de clase III se corresponden con los de mayor riesgo para el paciente.
Clase I
- Productos que no entran en contacto con el paciente o que entran en contacto solo con la piel intacta.
- Productos que penetran por un orificio corporal como la boca o nariz, de uso pasajero. A corto plazo los introducidos por cavidad oral hasta faringe, conducto auditivo externo hasta tímpano y cavidad nasal
- Productos destinados a ser utilizados como barrera mecánica, compresión o absorción de exudados.
Se excluyen de esta clase los productos que, aunque no entran en contacto con el paciente, pueden influir en procesos fisiológicos (productos que tratan la sangre destinada a reinfundirse) o los que suministran energía al cuerpo humano (equipos de radiodiagnóstico).
Ejemplos de productos sanitarios de esta clase son: bolsas de orina, vendas, medias elásticas, andadores, bastones y enemas.
En esta clase se incluyen también las siguientes subclases:
Clase I estériles
Son de la clase I con presentación estéril.
Ejemplos de estos son: guantes de examen estériles, jeringuillas, gasas estériles para proteger heridas.
Clase I con función de medición
Son de la clase I y sirven para medir.
Ejemplos de estos son: tonómetros o termómetros no electrónicos.
Clase IIa
- Productos que se introducen por orificio corporal o por medios quirúrgicos, a través de la piel, y no están destinados a permanecer en él.
- Productos no invasivos destinados a la conducción o almacenamiento de sangre, fluidos, o tejidos corporales, líquidos o gases destinados a una perfusión, administración introducción en el cuerpo humano
- Productos que modifican procesos fisiológicos siempre que no se efectué de forma potencialmente peligrosa.
- Productos que se utilizan el tratamiento de heridas no incluidos en las clases I y IIb.
- Productos que se introducen por orificio corporal o por medios quirúrgicos, a través de la piel, de uso a corto plazo. De uso prolongado los introducidos por cavidad oral hasta faringe, oído externo hasta tímpano y cavidad nasal que no pueden sr absorbidos por la membrana mucosa.
- Productos invasivos en orificios corporales, salvo los quirúrgicos, destinados a conectarse a producto sanitario activo de clase IIa o superior.
- De forma general los productos invasivos de tipo quirúrgico destinados a uso pasajero o corto plazo, excepto los indicados en la clase IIb y III
- Productos implantables y productos invasivos que se coloquen dentro de los dientes.
- Desinfectantes de productos no invasivos.
Ejemplos de productos de la clase IIa son: circuitos de circulación extracorpórea, sondas urológicas, drenajes quirúrgicos, agujas, catéteres, guantes quirúrgicos, lentes de contacto y esfigmomanómetros.
Clase IIb
- Productos no invasivos que se utilizan para influenciar los procesos fisiológicos o que administran sustancias o energía de forma potencialmente peligrosa y los destinados al diagnóstico de las funciones vitales.
- Productos no invasivos destinados a modificar la composición biológica o química de células o tejidos humanos, de la sangre, de otros líquidos corporales o de otros líquidos destinados a implantarse o administrarse en el cuerpo, salvo si el tratamiento para el que el producto se usa consiste en filtración, centrifugación o intercambios de gases o de calor.
- Productos no invasivos que se utilizan para la heridas con ruptura de la dermis y sólo pueden cicatrizar por segunda intención.
- Productos que se introducen por orificio corporal o por medios quirúrgicos, a través de la piel, de uso prolongado.
- Productos invasivos que se destinen para administrar energía al cuerpo humano o intercambiarla con el mismo de forma potencialmente peligrosa, teniendo en cuenta la naturaleza, la densidad y el punto de aplicación de la energía
- Todos los productos activos destinados a emitir radiaciones ionizantes con fines terapéuticos, incluidos los productos para controlar o supervisar dichos productos, o que influyan directamente en el funcionamiento de los mismos.
- Productos invasivos de uso pasajero para ejercer un efecto biológico o a ser absorbidas totalmente o en gran parte.
- Productos anticonceptivos o para prevención de enfermedades de transmisión sexual.
- Productos desinfectantes para productos invasivos y los de limpieza de lentes de contacto.
- Productos invasivos de uso a corto plazo excepto los que pertenecen a la clase IIa.
- Productos invasivos de uso prolongado plazo excepto los que pertenecen a la clase IIa y III.
- Productos implantables que no sean IIa o III.
Ejemplos de productos de la clase IIb son: lentes intraoculares, suturas quirúrgicas no absorbibles, apósitos para heridas que cicatrizan por segunda intención, bolsas de sangre, plumas de insulina, equipos de RX diagnósticos, máquinas de anestesia y sistemas de vigilancia de cuidados intensivos.
Clase III
- Productos invasivos destinados a entrar en contacto con el sistema nervioso central y el sistema circulatorio central para diagnóstico, control y tratamiento.
- Productos que se absorben totalmente y los que contienen sustancias medicinales.
- Los implantes mamarios y las prótesis articulares de cadera, rodilla y hombro
Ejemplos de productos de la clase III son: suturas absorbibles, adhesivos internos biológicos, endoprótesis vasculares (stents), prótesis de cadera, válvulas cardiacas y catéteres recubiertos de medicación.
3. MARCADO CE. ORGANISMOS NOTIFICADOS
El Marcado CE de conformidad es un requisito obligatorio para la comercialización de productos sanitarios en Europa. Desde 1998, ningún producto sanitario puede comercializarse en los países europeo sin este marcado según la Directiva 93/42/CEE, vigente hasta la entrada en vigor del Reglamento (UE)2017/745. Tiene un periodo de validez de 5 años y es prorrogable de 5 en 5 años. Se indica en su etiquetado e instrucciones de uso mediante el símbolo:

La identificación sin número es la autocertificación y con número se debe a que se realiza por un Organismo Notificado. Cada Organismo Notificado tiene un número distintivo.
Para poder venderse en el Espacio Económico Europeo, o por sus siglas, el EEE (formado por la UE más Islandia, Liechtenstein y Noruega), muchos productos deben llevar obligatoriamente el marcado CE, que constituye la prueba de que el producto se ha evaluado y cumple los requisitos de seguridad, sanidad y protección del medio ambiente exigidos por la UE. Es válido para los productos fabricados tanto dentro como fuera del EEE, cuya comercialización esté prevista dentro del mismo. A través del siguiente link se puede comprobar el índice de productos con distintivo CE; http://europa.eu/youreurope/business/product/ce-mark/index_es.htm
Los Organismos Notificados son los que emiten los certificados correspondientes, siguiendo los procedimientos establecidos para la evaluación de los productos autorizando la comercialización y realizan el seguimiento de la postcomercializacion. Estos certificados tienen que hacer mención a alguno de los Anexos de la Directiva que corresponda (Directiva 90/385/CEE, Directiva 93/42/CEE o Directiva 98/79/CE), vigentes hasta la entrada en vigor del Reglamento (UE) 2017/745 y Reglamento (UE) 2017/746.
El proceso para la obtención del certificado CE es:
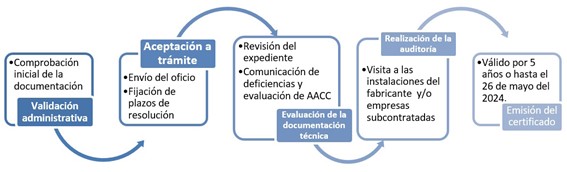
Fuente Organismo Notificado 0318. Informacion y condiciones para el marcado CE.
En España los organismos notificados son designados por las autoridades nacionales, que ostentan la capacidad de supervisión sobre las actividades de dichos organismos, debiendo éstos facilitarles toda la información necesaria, incluida la relativa a los certificados emitidos, rechazados, suspendidos o retirados. Está recogido en la Directiva 93/42/CEE del Consejo de Europeo, traspuesta al RD 414/1996 y posteriormente al RD 1591/2009 donde se detalla el procedimiento a seguir por el organismo notificado en sus actividades de certificación de la conformidad solicitadas por los fabricantes, hasta la entrada en vigor de los nuevos Reglamentos.
La designación del Organismo Notificado español fue realizada por las Autoridades Sanitarias Españolas en 1995, para los productos sanitarios y los productos sanitarios implantables activos, en diciembre del 2002, para los productos sanitarios para diagnóstico “in Vitro” y en el 2004, para los productos sanitarios que incorporan derivados de tejidos animales. Para todos ellos, el ámbito de la designación se extiende a todos los productos en cuya evaluación de conformidad debe intervenir un Organismo Notificado y para todos los procedimientos de evaluación de conformidad. El Organismo Notificado 0318 ha sido redesignado nuevamente en marzo de 2010.
Posteriormente, mediante el Real Decreto 1275/2011, de 16 de septiembre, se crea la Agencia estatal “Agencia Española de Medicamentos y Productos Sanitarios” y se aprueba su Estatuto. Entre sus competencias está:
“Actuar como Organismo Notificado, evaluando la conformidad de los productos sanitarios, realizando las auditorías de los sistemas de calidad, certificando las normas específicas de dichos sistemas y emitiendo los certificados CE con vistas a la colocación del marcado CE en dichos productos, en los términos queestablezca la designación efectuada por el Ministerio de Sanidad, Servicios Sociales e Igualdad , así como autorizar las entidades colaboradoras en la certificación de los productos sanitarios”
La intervención del organismo notificado depende de la clasificación de riesgo del producto y se evalúan distintos procesos, según se indica a continuación:
- PS Clase I: No requiere la intervención del Organismo Notificado, el fabricante realiza la autocertificación de marcado CE.
- PS Clase I estériles: La intervención del Organismo Notificado se limitará únicamente a los aspectos relacionados con la obtención y mantenimiento de las condiciones de esterilidad.
- PS Clase I con función de medición: La intervención del Organismo Notificado se limitará únicamente a los aspectos relacionados con la función de medición.
- PS Clase I estériles con función de medición: La intervención del Organismo Notificado se limitará a los aspectos relacionados con la obtención y mantenimiento de la esterilidad y con la función de medición.
- PS Clase IIa: La intervención del Organismo Notificado irá encaminada a evaluar el cumplimiento de todos los requisitos esenciales aplicables al producto. Fabricación y la esterilización.
- PS Clase IIb: La intervención del Organismo Notificado irá encaminada a evaluar el cumplimiento de todos los requisitos esenciales aplicables al producto. Diseño, fabricación, esterilización y ensayos clínicos.
- PS Clase III: La intervención del Organismo Notificado irá encaminada a evaluar el cumplimiento de todos los requisitos esenciales aplicables al producto. Aprobación previa de diseño, fabricación, esterilización y ensayos clínicos
4. ETIQUETADO E INSTRUCCIONES
Todos los productos sanitarios deben estar identificados según indica el RD. El fabricante indicará en cada producto la información necesaria para su utilización con plena seguridad, teniendo en cuenta la formación y los conocimientos de los usuarios potenciales y para identificar al fabricante.
La información constará de las indicaciones que figuren en la etiqueta y las que figuren en las instrucciones de utilización.
Los datos necesarios para la utilización del producto deberán figurar, cuando sea factible, en el propio producto y/o en un envase unitario o en el envase comercial. Si no es factible envasar individualmente cada unidad, estos datos deberán figurar en unas instrucciones de utilización que acompañen a uno o varios productos. Excepcionalmente, las instrucciones no serán necesarias en el caso de los productos de las clases I y IIa.
El marcado de cumplimiento de RD es CE con unas dimensiones establecidas, si el marcado debe disminuirse o aumentarse se realizará en proporción. Existen una serie de datos que deben aparecer siempre en las etiquetas de los productos, cuando sea apropiado podrán ser en forma de símbolo deben ajustarse a las normas armonizadas. Siempre debe incluir:
- Nombre del fabricante y distribuidor autorizado si el fabricante no tiene domicilio social en la CEE.
- Información para identificación del producto y contenido del envase.
- Si es estéril debe aparecer indicando método de esterilización.
- El número de lote de fabricación o de serie.
- Fecha de caducidad expresada con mes y año. En los productos activos el año de fabricación.
- Si es de un solo uso.
- En los productos a medida la indicación de ello “producto a medida”.
- Si es un producto solo para investigación clínica debe quedar reflejado “exclusivamente para investigaciones clínicas”.
- Condiciones de almacenamiento y conservación.
- Instrucciones especiales de utilización.
- Cualquier advertencia o precaución que deba adoptarse. Por ejemplo si tiene látex.
- Método de esterilización, si es preciso
- Indicación de que el producto contiene como parte integrante una sustancia derivada de la sangre humana, si es preciso.
- Marcado CE con las medidas establecidas en el RD, si cambia de tamaño deberá mantener la proporción. El requisito de dimensiones se puede no cumplir en caso de productos muy pequeños.
- Sistema UDI
El Sistema UDI, como ya hemos comentado es uno de los grandes cambios del Reglamento de la Unión Europea 745/2017 donde aparece por primera vez. Con este sistema de identificación se va a crear una base de datos europea de todos los PS EUDAMED.
En esta base de datos se podrá encontrar toda la información de los PS respecto a registro, certificados, vigilancia en el mercado europeo e investigación clínica. Para acceder a la base de datos se realizará mediante el número de identificación UDI.
UDI es un número de identificación del producto, que consta de dos partes:
- DI: parte estática es el principal identificador del modelo de producto, incluye el código de la empresa, dígitos que debe completar la empresa y un digito de control. Esta identificación es un prefijo que asocia la entidad emisora de códigos UDI.
- PI: parte dinámica. Identifica la unidad de producción del producto.
Tiene que llevar incluido el número de serie, lote, fecha de identificación y fabricación y/o caducidad.
Los formatos aprobados para la lectura de los códigos son código de barras lineal, Datamatrix o RFID, debe figurar en la etiqueta del producto y embalajes.
La parte de UDI-DI no es obligatorio que aparezca en el etiquetado, es la forma de acceder a la base de datos EUDAMED. Donde sí debe figurar es en todos los certificados y declaraciones de conformidad del producto.
La implantación del marcado UDI está recogida en el artículo 123 del Reglamento de PS con ampliación de plazo en el Reglamento (EU) 2020/561 y en el 113 en el Reglamento de PS de diagnóstico in vitro. Loa plazos son:
- Asignación de UDI y envío a EUDAMED 26 de mayo 2021
- Marcado UDI Clase III 26 de mayo 2021
- Marcado UDI Clase IIa y IIb 26 de mayo 2023
- Marcado UDI Clase I 26 de mayo de 2025.
Las instrucciones de forma general deben incluir los datos suficientes para la comprensión por el usuario:
- Los datos del etiquetado.
- La forma de utilización.
- Los posibles efectos secundarios.
- Precauciones a tomar en su utilización.
- Instrucciones de montaje (si es necesario).
- Mantenimiento (si es necesario).
- La fecha de publicación de la última revisión de las instrucciones.
Además, si es un producto reutilizable deberá aparecer los procedimientos de uso, limpieza, desinfección y esterilización, si es un producto sanitario de solo uso, los riesgos que supondría reutilizarlo y si es un producto con función de medición, el grado de precisión.
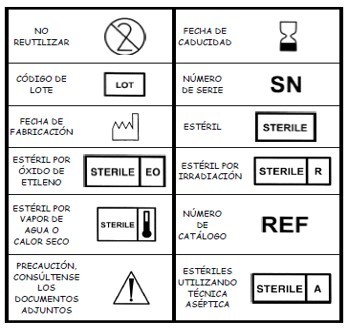
5. PICTOGRAMAS BÁSICOS
Los datos relacionados con los productos sanitarios pueden adoptar, cuando sea apropiado, la forma de símbolos. Los símbolos y los colores de identificación que se utilicen deberán ajustarse a las normas armonizadas. Si no existe ninguna norma al respecto, los símbolos y los colores se describirán en la documentación que acompañe al producto.
Los símbolos ajustados a normas armonizadas más utilizados para el etiquetado son:
BIBLIOGRAFIA
- RD 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios (BOE 6/11/2009).
- RD 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos. (BOE 6/11/2009).
- RD 1662/2000, De 29 de septiembre, sobre productos sanitarios para diagnóstico In vitro. (BOE 30/09/2000).
- RD 1193/2012 por el que se modifica 1662/2000 del 29 de septiembre, sobre productos sanitarios para diagnóstico In vitro.
- Directiva 93/42/CEE del Consejo, relativa a los productos sanitarios
- RD 1591/2009, de 16 de octubre, por el que se regulan los productos sanitarios (BOE 6/11/2009)
- RD 1616/2009, de 26 de octubre, por el que se regulan los productos sanitarios implantables activos. (BOE 6/11/2009)
- RD 1662/2000, De 29 de septiembre, sobre productos sanitarios para diagnóstico In vitro. (BOE 30/09/2000)
- RD 1193/2012 por el que se modifica 1662/2000 del 29 de septiembre, sobre productos sanitarios para diagnóstico In vitro.
- Mompart Garcia MP., Tourne Izquierdo BM., La gestión de los productos materiales del libro Actualizaciones Enfermería S21, Madrid; Ed. DAE (Grupo PARADIGMA), 2012. ISBN 978-84-92815-49-4.
- Ayuso Murillo D., Grande Sellera RF., La gestión de enfermería y los servicios generales en las organizaciones sanitarias, Madrid; Ed. Díaz de Santos, 2006 ISBN 84-7978-756-2.
- Ayuso Murillo D., Recursos Materiales. Curso en gestión de los Servicios de Enfermería. Madrid. Uned, 2007
- http://guiasjuridicas.wolterskluwer.es
- https://contratacionpublica.wikispaces.com/Pliegos+de+cláusulas+administrativas+gene