3.1 CONCEPTOS BÁSICOS
La farmacocinética estudia el recorrido que realiza un fármaco en el organismo desde su administración hasta su eliminación. Este proceso dinámico determina la concentración plasmática del fármaco en el tiempo, lo que a su vez condiciona su eficacia terapéutica y la aparición de posibles efectos adversos.
La comprensión de la farmacocinética es esencial para el personal sanitario, ya que permite ajustar dosis, elegir la vía de administración adecuada y anticipar interacciones o acumulaciones peligrosas, especialmente en situaciones críticas como las urgencias, el embarazo o en pacientes con insuficiencia renal o hepática.
CURVA DE CONCENTRACIÓN PLASMÁTICA VS TIEMPO
Uno de los pilares de la farmacocinética es la curva de concentración plasmática vs. tiempo, que refleja cómo el fármaco entra, se distribuye, se metaboliza y finalmente se elimina del cuerpo, es decir, refleja la concentración del fármaco en sangre en función del tiempo transcurrido.
Los principales parámetros que describen cuantitativamente la evolución de las concentraciones del fármaco en el organismo serían:
- Inicio de acción: momento en que el fármaco alcanza concentraciones efectivas.
- Concentración máxima (Cmax): nivel más alto de fármaco en sangre.
- Tiempo hasta la concentración máxima (Tmax): indica la velocidad de absorción del fármaco.
- Área bajo la curva (AUC): refleja la biodisponibilidad total.
- Vida media (t½): Es el tiempo necesario para que la concentración plasmática del fármaco se reduzca a la mitad. Determina la frecuencia de administración, la acumulación y el tiempo requerido para alcanzar el estado de equilibrio.
Ejemplo: La adenosina, empleada en taquicardias paroxísticas, tiene una vida media de segundos, lo que justifica su administración en bolo IV rápido.
- Concentración mínima eficaz (CME) y concentración tóxica, son las que delimitan el margen terapéutico.
- Concentración mínima eficaz (CME): valor por encima del cual el fármaco empieza a producir un efecto terapéutico.
- Concentración tóxica: nivel a partir del cual comienzan a aparecer efectos adversos significativos.
- Margen o ventana terapéutica: también llamado, intervalo terapéutico. Es el rango comprendido entre la concentración mínima eficaz y la concentración tóxica de un fármaco. Es decir, el rango de concentraciones plasmáticas de un fármaco en el cual es efectivo sin ser tóxico. Si un fármaco tiene un margen estrecho implica mayor riesgo de toxicidad y necesidad de monitorización estrecha.
Ejemplo: La digoxina requiere control plasmático frecuente debido a su estrecho margen terapéutico, con riesgo de arritmias por sobredosificación. - Periodo de latencia: es el tiempo que transcurre desde que se comienza a administrar el fármaco hasta que este alcanza la CME. Para evitar o disminuir ese periodo podemos administrar el fármaco por vía intravenosa directamente a la concentración eficaz.
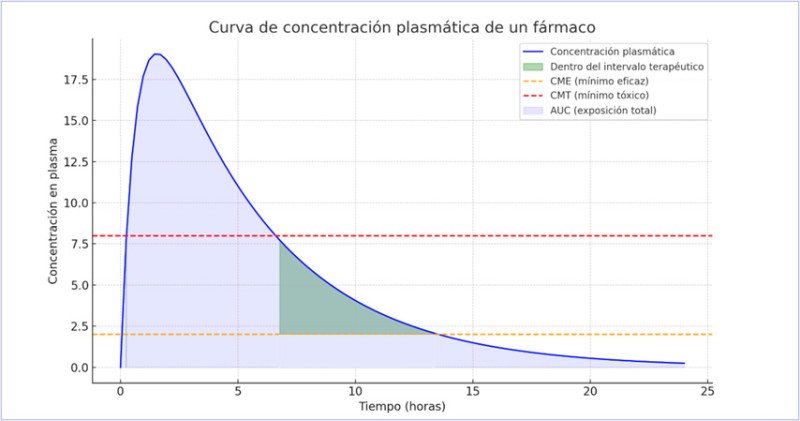
Imagen 2.
- Biodisponibilidad: Es el porcentaje de la dosis administrada de un fármaco que alcanza la circulación sistémica en forma activa. Depende de la vía de administración, de los procesos de absorción y del metabolismo hepático de primer paso.
Ejemplo: El paracetamol oral tiene una biodisponibilidad de aproximadamente 60–70%, mientras que su forma intravenosa alcanza una biodisponibilidad del 100%.
- Volumen de distribución (Vd): Describe cómo un fármaco se distribuye en el organismo después de ser absorbido o administrado. Su valor depende tanto de las propiedades del fármaco como de las características del paciente.
- Vd bajo: el fármaco permanece principalmente en el plasma (no se distribuye mucho).
Ejemplo: La aspirina IV u oral en síndrome coronario aguda, tiene un bajo Vd por tanto se mantiene en sangre y actúa rápidamente sobre plaquetas circulantes. Es ideal para un control rápido de agregación plaquetaria.
- Vd alto: el fármaco se distribuye ampliamente en tejidos y órganos, incluso fuera del sistema vascular.
Ejemplo: El diazepam tiene un Vd alto, se distribuye ampliamente en los tejidos, incluyendo el sistema nervioso central (SNC). Su alta liposolubilidad hace que atraviese con facilidad la barrera hematoencefálica y se acumule en tejido graso. Esto explica su inicio rápido y su larga duración de acción, especialmente cuando se administra IV.
- Dosis de carga y dosis de mantenimiento
- Dosis de carga: cantidad inicial de fármaco administrada para alcanzar rápidamente concentraciones plasmáticas terapéuticas.
- Dosis de mantenimiento: cantidad periódica administrada para mantener dichas concentraciones en el tiempo.
Ejemplo: En estado epiléptico, se puede administrar una dosis de carga de fenitoína (15–20 mg/kg IV), seguida por dosis de mantenimiento ajustadas al nivel plasmático y función hepática.
3.2 PROCESO LADME
El conocimiento de la farmacoterapia y fluidoterapia es la base del manejo del paciente tanto en urgencias como en cualquier ámbito hospitalario. Para su correcto manejo, no solo se deben tener las habilidades técnicas necesarias para la administración, sino también conocimientos sobre farmacocinética.
Ésta comprende los siguientes procesos: liberación, absorción, distribución, metabolismo y excreción del fármaco (LADME).
3.2.1 Liberación
Es el proceso mediante el cual el principio activo es liberado del fármaco, para, posteriormente, disolverse y absorberse en el torrente sanguíneo. No todos los fármacos pasan por el proceso de liberación, esto depende de la vía de administración y la forma farmacéutica.
- Ejemplo: Los fármacos que se presentan en forma líquida y se administran por vía oral no necesitan liberarse, pues se absorben inmediatamente, los fármacos nebulizados o administración intravenosa, tampoco, en cambio un fármaco de administración oral en cápsula sí que lo precisaría.
3.2.2 Absorción
Es el proceso mediante el cual el fármaco, una vez liberado (si lo ha precisado) de la forma farmacéutica en la que se administra, llega hasta el torrente sanguíneo.
La absorción, por tanto, es el paso a través de membranas celulares.
Depende de la liposolubilidad y el grado de ionización, así́ como de las características de la preparación farmacéutica, del lugar de absorción y de los fenómenos de eliminación presistémica como, por ejemplo, el primer paso hepático.
Entre los factores que alteran la absorción pueden hallarse factores fisiológicos, como la edad, el embarazo o la presencia de alimentos; factores patológicos, como enfermedades que cursan con diarrea, vómitos o alteraciones de la absorción, y factores iatrogénicos debidos a interacciones o incorrecta administración de los preparados.
3.2.3 Distribución
En este proceso el fármaco, ya absorbido, se reparte por la sangre a todo el cuerpo atravesando las membranas de los capilares hacia los tejidos. En la sangre el fármaco puede ir:
- Unido a proteínas plasmáticas: la mayoría de las veces, el fármaco se une a proteínas que están en el plasma sanguíneo, principalmente a la albúmina. Esta unión es reversible y actúa como un "depósito" o reserva del fármaco. Cuando el fármaco está unido a estas proteínas, no puede atravesar las membranas de los capilares ni ejercer su efecto farmacológico.
- Disuelto como fracción libre: es la parte del fármaco que no está unida a proteínas plasmáticas, está libre y disuelta en el plasma. Solo esta fracción libre puede: salir de la sangre hacia los tejidos, atravesando las membranas capilares y ejercer el efecto farmacológico porque puede interactuar con las células y receptores en los tejidos, puede también ser metabolizada o eliminada del organismo.
Ejemplo: El diazepam, es un fármaco que se une fuertemente a proteínas, esta unión a proteínas plasmáticas (como la albúmina) actúa como un “reservorio” del fármaco en sangre, aunque solo la fracción libre actúa, el fármaco se va liberando progresivamente desde la fracción unida lo que prolonga su efecto.
En situaciones de hipoalbuminemia (embarazo, hepatopatía, desnutrición): ↓ albúmina → ↑ fracción libre → ↑ riesgo de toxicidad en fármacos con alta unión proteica (ej. fenitoína, diazepam).
El desplazamiento y la distribución de los fármacos hasta los distintos órganos dependen de su vascularización. Así, en órganos como el corazón, el hígado y los riñones, pueden encontrarse concentraciones de fármacos más elevadas que en zonas del organismo poco vascularizadas, como el tejido celular subcutáneo.
3.2.4 Metabolización o biotransformación
Es el proceso mediante el cual el organismo transforma los fármacos en compuestos más sencillos (metabolitos), generalmente menos activos y más fáciles de eliminar.
Este proceso ocurre sobre todo en el hígado y tiene como finalidad inactivar el fármaco o facilitar su eliminación. En algunos casos, los metabolitos son activos o incluso necesarios para que el fármaco tenga efecto (como los profármacos).
- Ejemplo: El diazepam se metaboliza en el hígado y forma metabolitos activos. Por eso puede tener un efecto prolongado en pacientes añosos o con insuficiencia hepática.
El clopidogrel es un profármaco que necesita metabolizarse para ejercer su acción antiplaquetaria.
Un concepto importante por comprender es el llamado “metabolismo de primer paso que es el fenómeno por el cual un fármaco administrado por vía oral es metabolizado significativamente en el hígado (o en la mucosa intestinal) antes de llegar a la circulación sistémica. Esto reduce su biodisponibilidad.
Evitar este primer paso hepático (como ocurre en la vía sublingual, en la rectal de forma parcial, en la intravenosa o en la inhalada) permite: un inicio de acción más rápido, menor pérdida de fármaco, dosis más bajas para lograr efecto terapéutico, mayor eficacia clínica.
- Ejemplo: La nitroglicerina presenta un metabolismo hepático de primer paso tan intenso que se inactiva casi completamente si se administra por vía oral. Por ello, se utiliza por vía sublingual para un efecto rápido.
Es importante saber que diversos factores pueden influir en la actividad metabólica hepática, incrementando así el riesgo de interacciones farmacológicas y potenciales toxicidades.
Entre los factores más habituales están la edad, los inductores enzimáticos ( fármacos o sustancias que aumentan la actividad metabolizarte , originando una disminución de la intensidad o la duración del efecto del fármaco), los inhibidores enzimáticos (pueden reducir o inhibir el metabolismo de otro fármaco cuando ambos se metabolizan por sistemas enzimáticos comunes, la consecuencia clínica será un incremento de la semivida del fármaco cuyo metabolismo es inhibido, lo cual puede conducir a un aumento de su actividad farmacológica ) y factores patológicos como la insuficiencia hepática.
- Ejemplo: El Ketoconazol (antifúngico) es un fármaco inhibidor que afecta a la enzima hepática CYP3A4 (citocromo P450 3A4), el midazolam y el fentanilo también se metabolizan por esta enzima. Al administrar ketoconazol junto con midazolam o fentanilo, la inhibición del CYP3A4 disminuye el metabolismo de estos fármacos y esto aumenta la concentración plasmática de midazolam o fentanilo, lo que puede llevar a una sedación excesiva, depresión respiratoria o toxicidad.
La fenitoína y la rifampicina son potentes inductores de enzimas hepáticas del sistema citocromo P450, especialmente CYP3A4 y otras isoenzimas. Tanto la warfarina como el fentanilo se metabolizan, al menos en parte, a través de estas vías enzimáticas. Por ello, la coadministración de fenitoína o rifampicina puede acelerar el metabolismo de estos fármacos, reduciendo su concentración plasmática y disminuyendo su eficacia terapéutica. Esta interacción puede conducir a un aumento del riesgo trombótico debido a la subdosificación de warfarina o a un control insuficiente del dolor en pacientes tratados con fentanilo."
3.2.5 Excreción
Es la eliminación del fármaco sin modificar o en forma de metabolitos. Las vías de eliminación más importantes son: renal (a través de la orina), digestiva (a través de las heces) y biliar (a través de la bilis).
En caso de que el fármaco se concentre en el órgano excreto, podría causar toxicidad en este. Cuando el órgano excretor presenta alguna alteración, el fármaco podría prolongar su acción en el organismo, debido a que permanecería en este durante un mayor tiempo.
VÍAS DE EXCRECIÓN:
- Vía gastrointestinal: incluye la excreción gástrica, por saliva, intestinal (a través de las heces) y biliar.
- Excreción biliar: es una de las rutas más relevantes. El fármaco o sus metabolitos son secretados por los hepatocitos hacia la bilis, y de ahí al intestino delgado. Algunos pueden ser reabsorbidos, lo que da lugar al ciclo enterohepático, prolongando su vida media y efecto farmacológico.
- Excreción a través del sudor: se elimina por esta vía el alcohol, las sulfamidas y la urea.
- Excreción por pelo y piel: utilizada sobre todo para el estudio en pruebas toxicológicas.
- Excreción lacrimal: se debe tener en cuenta a la hora de la administración de pomada oftálmicas que el iodo se elimina a través de las lágrimas. Si estas pomadas contienen mercurio, en presencia de iodo se formaría ioduro mercúrico, que es un compuesto altamente irritante.
- Excreción mamaria: de especial importancia en períodos de lactancia, ya que los metabolitos excretados pueden ser transmitidos al lactante.
- Excreción renal: El riñón es el principal órgano excretor del organismo, expulsando a través de la orina los metabólicos del fármaco. En el riñón la excreción se lleva a cabo por:
- Filtración glomerular de las moléculas con un peso molecular menor de 70.000 Dalton y que se encuentren libres en el plasma.
- Secreción tubular.
El aclaramiento renal es un concepto que nos indica cuánto volumen de plasma se limpia o "depura" completamente de un fármaco en un minuto a través del riñón. Se mide en mililitros por minuto (ml/min).
Para entender la eliminación renal total de un fármaco, debemos considerar tres procesos:
- Filtración glomerular: El fármaco pasa del plasma a la orina a través de los glomérulos del riñón. La cantidad filtrada depende de la velocidad de filtración glomerular (que es fija para cada persona) y de la cantidad de fármaco libre en plasma (el que no está unido a proteínas).
- Secreción tubular activa: Algunas sustancias pueden ser activamente transportadas desde la sangre hacia los túbulos renales para ser eliminadas. Esto puede aumentar la eliminación del fármaco más allá de lo que se filtra pasivamente.
- Reabsorción tubular: Algunos fármacos pueden ser reabsorbidos desde la orina hacia la sangre en los túbulos, disminuyendo su eliminación. Por eso, para conocer la eliminación renal total, se resta la cantidad reabsorbida.
Una vez comprendido el mecanismo de eliminación renal de los fármacos, es fundamental destacar que el pH de la orina puede influir significativamente en la eliminación de ciertos compuestos, especialmente en situaciones clínicas críticas como las intoxicaciones agudas.
En urgencias, la modificación del pH urinario es una estrategia terapéutica utilizada para aumentar la solubilidad y reducir la reabsorción tubular de determinados fármacos o tóxicos, favoreciendo así su eliminación renal.
- Ejemplo clínico: Intoxicación por ácido acetilsalicílico.
Los salicilatos son ácidos débiles. En medio alcalino, se ionizan más y no pueden atravesar fácilmente la membrana tubular renal, lo que favorece su excreción urinaria y disminuye su reabsorción. Este principio se conoce como "ion trapping" (atrapamiento iónico), utilizado como estrategia de eliminación forzada.
Paciente de 30 años, ingesta autolítica de ácido acetilsalicílico, 40 comprimidos de 500 mg hace 3 horas.
Laboratorio: salicilatemia 48 mg/dL, pH arterial 7.30, hiperventilación compensadora.
Tratamiento en urgencias: fluidoterapia con suero glucosado, bicarbonato sódico IV para alcalinizar la orina, objetivo pH urinario 7.8. Se corrige hipopotasemia leve. Se evita diálisis. Evolución favorable.
3.3 CARACTERÍSTICAS FARMACOCINÉTICAS RELACIONADAS CON LA VÍA DE ADMINISTRACIÓN
Como es de esperar, la vía de administración de un fármaco determinará el modo en que éste accede al organismo y, por tanto, influye directamente en su farmacocinética.
Comprender estas diferencias es fundamental en contextos clínicos como las urgencias, donde la selección de la vía más adecuada puede ser clave para lograr un efecto rápido, seguro y eficaz.
VÍA INTRAVENOSA (IV)
- Características: Administración directa al torrente sanguíneo → absorción inmediata.
- Concentración plasmática: Aumenta bruscamente hasta un pico máximo (Cmáx) inmediato y luego desciende progresivamente por eliminación.
- Ventajas: Acción rápida, útil en situaciones críticas.
Ejemplo:administración de fentanilo intravenoso para control del dolor en paciente politraumatizado.
VÍA ORAL (V.O)
- Características: Requiere absorción a través del tracto gastrointestinal → inicio más lento.
- Concentración plasmática: Pico moderado, alcanzado más tarde; mayor variabilidad por el metabolismo de primer paso hepático.
- Ventajas: Fácil administración, pero menos útil en urgencias agudas.
Ejemplo: ondansetrón oral en niños con vómitos que no precisan vía intravenosa.
VÍA SUBCUTÁNEA (SC)
- Características: El fármaco se deposita en el tejido subcutáneo, desde donde se absorbe lentamente. Tiene menor vascularización que el músculo, por lo que la absorción es más lenta que por vía IM.
- Concentración plasmática: Pico más tardío, con menor intensidad, pero sostenido en el tiempo. La absorción puede verse afectada por el flujo sanguíneo (shock, frío).
- Ventajas: Ideal para fármacos que requieren liberación prolongada o cuando se quiere evitar el acceso venoso. No apta para emergencias con necesidad de efecto rápido.
Ejemplo : morfina sc para el manejo del dolor en cuidados paliativos o urgencias crónicas sin acceso IV.
VÍA INTRAMUSCULAR (IM)
- Características: El fármaco se administra directamente en el músculo, desde donde se absorbe al torrente sanguíneo. La absorción es más rápida que por vía oral, pero más lenta que la intravenosa.
- Concentración plasmática: Alcance del pico en unos 5–10 minutos, con efecto clínico rápido. La velocidad depende del flujo sanguíneo muscular y la formulación del fármaco.
- Ventajas: Útil en situaciones sin acceso venoso, en pacientes agitados, niños o en entornos extrahospitalarios. Requiere menos habilidad técnica que la vía IV.
Ejemplo: ketamina im sedación rápida en pacientes agitados o para procedimientos dolorosos sin acceso venoso.
VÍA RECTAL
- Características: El fármaco se administra por el recto, donde se absorbe a través de la mucosa rectal. Absorción irregular. Una parte evita el metabolismo hepático de primer paso, lo que mejora su biodisponibilidad frente a la vía oral.
- Concentración plasmática: Variable y menos predecible, pico intermedio. Inicio más lento que IM o IV, pero útil si no se puede administrar por otra vía.
- Ventajas: Útil en niños, pacientes inconscientes, con vómitos o sin acceso venoso. No requiere técnica estéril.
Ejemplo: diazepam rectal en convulsiones de paciente pediátrico.
Tabla resumen de parámetros farmacocinéticos por vía de administración
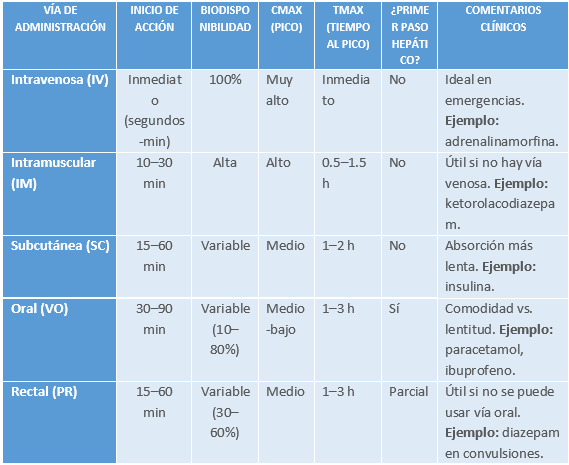
Tabla 2.
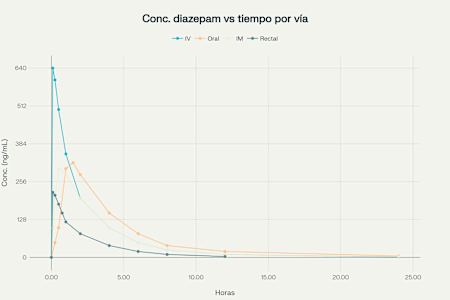
Imagen 3. Gráfica simulación de la absorción diazepam en vía intravenosa), oral, intramuscular (IM) y rectal.
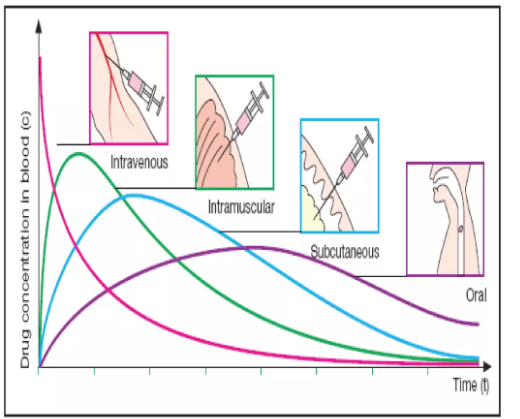
Imagen 4.
BIBLIOGRAFÍA
- Adams MP. Farmacología para enfermería. Un enfoque fisiopatológico. Pearson. 2009.
- Bodenham A. Acceso Vascular. Rev Médica Clínica Las Condes. 2017;28(5):713-26.
- Brunton L. Las bases farmacológicas de la terapéutica. 11ºed. McGraw-Hill. México. 2005.
- Flórez J. Farmacología humana. 6ª ed. Elsevier. 2014
- Hopfer Deglin J. Guía farmacológica para profesionales de enfermería. McGraw-Hill.
- Katzung BG. Farmacología básica y clínica. 11ª ed. McGraw-Hill. 2010.
- Mendoza PN. Farmacología médica. 1ª ed. Panamericana. México. 2008
- Mosquera JM. Farmacología clínica para enfermería. 3ªed. McGraw-Hill.2012
- Myeck MJ. Farmacología. 2ª ed. McGraw-Hill. México. 2004
- Pierre MAC. Manuel de farmacología básica y clínica. 5ª ed. McGraw-Hill.México. 2010.
- Pérez González A. Sociedad Española de Urgencias de Pediatría. [Internet]Administración de fármacos por vía intranasal. 2021(1). Disponible en https://seup.org/1-jornada-enfermeria-urgencias-pediatricas/procedimientos-de-enfermeria/
- Rang HP. Farmacología. 5º ed. Elsevier. 2004.
- Rodríguez CR. VAM Vademécum académico de medicamentos. 4ª. Ed. México. 2005.
- Rodríguez Palomares C. Farmacología para enfermeras. McGraw-Hill.2011
- Tiziani A. Harvard. Fármacos en enfermería. 4ª ed. Manual Moderno. 2011
- Trejo FCS. Fundamentos de farmacología. 1ª ed. McGraw-Hill.México. 2009
- Velázquez. Manual de farmacología: básica y clínica. Paramerica. 2014
- Agencia Española de Medicamentos y Productos Sanitarios (AEMPS). Fichas técnicas y alertas. https://cima.aemps.es
- ISMP-España. Errores de Medicación y Medicamentos de Alto Riesgo. https://www.ismp-espana.org
- Organización Mundial de la Salud. Estrategia Mundial sobre Seguridad del Paciente 2021–2030.
- Ministerio de Sanidad. Estrategia de Seguridad del Paciente del Sistema Nacional de Salud.
- Surviving Sepsis Campaign (2021)
- PHTLS (10ª ed.).