20.1 DEMENCIA: GENERALIDADES
20.1.1 Concepto y epidemiología
La demencia es un síndrome clínico, generalmente progresivo, que se da en varias enfermedades. En la demencia se ven afectadas las funciones cognitivas como la memoria, las praxias y las funciones ejecutivas, pero también afectan al comportamiento e interfieren en la capacidad de relacionarse y de llevar una vida normal.
La prevalencia de la demencia es mayor cuanto mayor sea el grupo de edad. En España, en personas entre los 40 y los 65 años la prevalencia se estima que está en torno al 0,05 % y va en aumento hasta situarse en el 39 % en el grupo de mayores de 90 años.
Con el envejecimiento de la población española, se estima que en 2050 se alcancen los 2 millones de personas con algún tipo de demencia diagnosticada.
20.1.2 Etiología
La demencia puede tener muchos orígenes; por ejemplo, enfermedades degenerativas en las que la manifestación principal es la demencia (demencias primarias), enfermedades en las que aparece la demencia en la última etapa del proceso como parte del cuadro clínico (enfermedad de Parkinson), demencias secundarias debido a déficits, tumores o tóxicos, y las demencias ocasionadas por accidentes cerebrovasculares.
La demencia no es fisiológica; siempre será patológica. El término demencia senil está totalmente fuera de lugar porque el envejecimiento, per se, no produce demencia.
La mayor parte las demencias son irreversibles; sin embargo, hay algunas que sí pueden revertirse como el SCA o la pseudodemencia por depresión.
A continuación, se ofrece una imagen a modo de esquema:
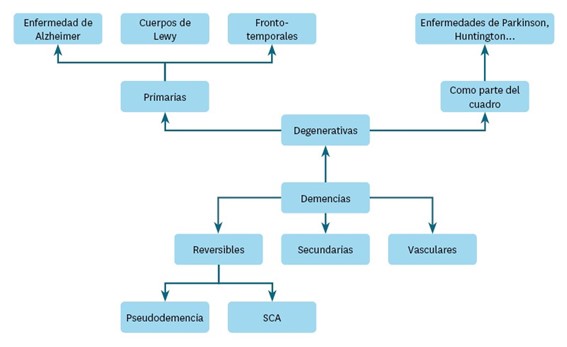
Imagen 12. Clasificación general de las demencias
20.1.3 Clínica
Las manifestaciones clínicas de la demencia son heterogéneas porque la etiología es muy diversa. Además, dependiendo de si son progresivas o no y de qué zonas anatómicas estén afectadas, la sintomatología será diferente. La demencia en general afecta a la memoria, el pensamiento, la orientación, la comprensión, el cálculo, la capacidad de aprendizaje, el lenguaje y el juicio. Las manifestaciones comunes utilizadas para identificarla son las siguientes:
- Deterioro cognitivo: es el resultado esencial de las demencias y lo que compromete más la dependencia. Entre los problemas cognitivos, a su vez, destacan estos:
o Amnesia: pérdida de memoria. Se da en casi todo tipo de demencias. Algunos autores lo consideran esencial, pero hay demencias que no lo desarrollan hasta las últimas etapas (p. ej.: la enfermedad de Parkinson).
o Afasia: incapacidad de expresarse.
o Agnosia: incapacidad de reconocer objetos o personas mediante uno o varios sentidos.
o Apraxia: incapacidad de realizar tareas que requieren recordar patrones o secuencias de movimientos a pesar de tener la capacidad física para hacerlo. Por ejemplo: no poder atarse los botones, aunque físicamente sí podría.
o Diminución de las funciones ejecutivas: razonamiento, capacidad de planificar, secuenciar, pensamiento abstracto.
- Cambios en el temperamento o la personalidad: aparecen en distinto grado y en distinto momento en función de la enfermedad.
- Síntomas conductuales y psicológicos: serie de síntomas relacionados con la alteración de la percepción, del contenido del pensamiento, del ánimo y de la conducta que pueden presentarse en las personas afectadas de demencia, y que constituyen parte de la expresión de la enfermedad:
o Trastornos del estado de ánimo (depresión, ansiedad y apatía).
o Agitación (agresividad, irritabilidad, inquietud, gritos y deambular errático).
o Síntomas psicóticos (alucinaciones visuales, auditivas y delirios).
En cuanto al curso evolutivo, las demencias de origen neurodegenerativo tienen en común un comienzo habitualmente insidioso y un curso evolutivo progresivo. En esta evolución también influye la capacidad de reserva, los mecanismos compensatorios desarrollados por el enfermo y el estado de las estructuras sociales que dan soporte.
En la mayoría de criterios diagnósticos (DSM, CIE...) consideran que los síntomas deben durar más de 6 meses.
20.2 ENFERMEDAD DE ALZHEIMER
20.2.1 Concepto y epidemiología
La enfermedad de Alzheimer (EA) es una patología primaria degenerativa y de evolución progresiva donde destaca el deterioro cognitivo y la demencia. El síntoma principal es la pérdida de memoria gradual y continua.
La EA es la causa principal de las demencias; en concreto, es responsable del 60 % o 70 % de todas ellas.
Igual que en el resto de demencias, el riesgo de desarrollar la EA va aumentando con la edad.
20.2.2 Etiología
Aunque la etiología exacta de la EA es desconocida, se postula una interacción entre factores genéticos, ambientales y de estilo de vida. Las hipótesis actuales giran en torno a la acumulación de β-amiloide y proteína tau. Los principales factores de riesgo incluyen:
- La vejez: cuanto más mayores, mayor incidencia.
- Antecedentes familiares: aunque no se considera una enfermedad hereditaria, sí se ha visto que en algunas familias la prevalencia es mayor.
- Síndrome de Down: mayor riesgo y aparición más temprana.
- Traumatismos craneales: parece haber relación con la aparición de la EA.
20.2.3 Clínica
- En las pruebas de imagen, destacan el aumento del número de ovillos neurofibrilares y placas amiloides (placas seniles).
- Amnesia: inicialmente sutil que provoca la incapacidad de retener nueva información. A medida que avanza la enfermedad, afectará a todos los tipos de memoria.
- Desorientación temporal:en un inicio, junto a la pérdida de memoria, aparecen algunos episodios de desorientación en el tiempo. El paciente suele negar estos episodios, aunque es consciente de ellos. La desorientación en espacios conocidos se da en fases más avanzadas.
- Anomia:incapacidad para nombrar objetos (cuesta encontrar las palabras), se da en la fase inicial junto con la amnesia.
- Apraxia, afasia y agnosia:en fases más evolucionadas aparecen estos signos.
- Funciones ejecutivas alteradas.
- Síntomas psicológicos y conductuales:en fases avanzadas es muy común. Muchas veces pueden ser causadas por dolor, malestar general, aburrimiento, desorientación, depresión...
- Aumento progresivo de la dependencia.
La clínica varía en función de la etapa evolutiva de la enfermedad y, debido a la inmensa heterogeneidad de las personas, es bastante complicado realizar una clasificación uniforme. Aun así, la escala de deterioro global de Reisberg (Global Deterioration Scale, GDS) hace una clasificación de los síntomas cognitivos y conductuales en 7 tramos diferentes. Reisberg realizó también la Functional Assessment Staging (FAST) para definir los cambios funcionales de la EA.
Para que sirva de pista, Reisberg afirma que la EA tiene una evolución de retrogénesis, es decir, que las etapas van devolviendo a la persona hasta la fase de neonato. Así, en la etapa 1 apenas hay afectación, pero en el peor caso de la etapa 7 estarán los pacientes en posición fetal con reflejo de succión y sin expresión.
La evolución es variable, pero la esperanza de vida media tras el diagnóstico suele situarse entre 8 y 12 años, pudiendo prolongarse hasta 15-20 en algunos casos.
20.2.4 Diagnóstico
No existe un diagnóstico certero para la EA; en general, se considera que hay EA si ocurren algunos de estos factores:
- Alteración de la memoria + afasia o apraxia o agnosia o alteración de funciones ejecutivas.
- Las alteraciones producen dificultades laborales y sociales.
- Instauración gradual y deterioro continuo.
- Las alteraciones no son atribuibles a otra cosa: trastornos neurológicos, SCA, alteración psiquiátrica...
La neuroimagen (TAC, RMN) y los biomarcadores (β-amiloide, tau) ayudan a confirmar el diagnóstico y a excluir otras demencias. La escala GDS de Reisberg permite valorar la etapa evolutiva.
20.2.5 Tratamiento y cuidados
Existen fundamentalmente 2 tipos de tratamiento farmacológico para la EA:
- Inhibidores de la enzima acetilcolinesterasa (IACE, por sus siglas en inglés): donepezilo, rivastigmina y galantamina.
- Memantina.
En cuanto a la terapia no farmacológica, destacan los siguientes cuidados:
- Cuidados básicos en función de la etapa y dependencia.
- Intervención o estimulación cognitiva: potencian, mantienen o recuperan capacidades cognitivas y mejoran el desempeño de AVD; también ayudan a socializar. P. ej., la reminiscencia, la orientación a la realidad, la estimulación sensorial...
- Cuidados en fases de angustia o agitación: similares a los del SCA.
- Cuidados al cuidador: información veraz y consejos para el autocuidado.
20.3 ENFERMEDAD DE CUERPOS DE LEWY
20.3.1 Concepto y epidemiología
La demencia por cuerpos de Lewy (CL) es una enfermedad neurodegenerativa asociada a la aparición de cuerpos de Lewy a nivel cortical y subcortical. Las características principales son los signos extrapiramidales, fluctuaciones cognitivas y alucinaciones visuales (Garzón-Giraldo, Montoya-Arenas, Carvajal-Castrillón, 2015).
La enfermedad o demencia por cuerpos de Lewy supone la 3.ª causa de demencia y representa el 10-15 % de todas ellas. Tras el diagnóstico tiene una media de 5 a 8 años de supervivencia.
20.3.2 Etiología
La etiología exacta es desconocida, aunque se asocia a la acumulación anómala de proteínas alfa-sinucleína y, en muchos casos, a depósitos concomitantes de beta-amiloide. Se ha descrito cierta agregación familiar, pero la mayoría de los casos son esporádicos. La edad avanzada es el principal factor de riesgo.
20.3.3 Clínica
La DCL combina síntomas de las enfermedades de Alzheimer y Parkinson, con una evolución fluctuante. Sus síntomas cardinales son:
- Fluctuaciones cognitivas: variaciones marcadas en la atención, la lucidez y el rendimiento mental.
- Alucinaciones visuales: recurrentes, bien estructuradas y vividas con gran realismo.
- Síntomas extrapiramidales (parkinsonismo): rigidez, bradicinesia y alteración de la marcha, habitualmente con afectación simétrica y escaso temblor de reposo.
- Trastorno de conducta del sueño REM: comportamientos motores anómalos (hablar, golpear o moverse violentamente durante el sueño) por pérdida de la atonía muscular propia de esta fase.
Otros síntomas frecuentes son los trastornos autonómicos (hipotensión ortostática, incontinencia, estreñimiento), los delirios sistematizados y las caídas repetidas.
En general, el deterioro cognitivo afecta precozmente a la atención y a las funciones visuoespaciales, mientras que la memoria suele estar menos comprometida en las fases iniciales.
20.3.4 Diagnóstico
El diagnóstico requiere la presencia de demencia (deterioro cognitivo suficiente para interferir en la vida diaria) junto con los criterios clínicos de la DCL.
Según el DLB Consortium (2017):
- Diagnóstico de DCL probable:
o Dos o más síntomas cardinales, o bien
o Un síntoma cardinal acompañado de al menos un biomarcador indicativo.
- Diagnóstico de DCL posible:
o Un solo síntoma cardinal sin biomarcadores, o
o Un biomarcador indicativo sin síntomas cardinales.
Biomarcadores indicativos:
- Disminución de la captación dopaminérgica en el DAT-SPECT o PET cerebral.
- Disminución de la captación miocárdica en la gammagrafía con MIBG.
- Polisomnografía que demuestra ausencia de atonía en el sueño REM.
En el diagnóstico es importante “La regla del año”. Esta regla ayuda a diferenciar la demencia con cuerpos de Lewy de la demencia asociada a la enfermedad de Parkinson.
- Si los síntomas motores del Parkinson aparecen más de 1 año antes que el deterioro cognitivo → Demencia asociada a la enfermedad de Parkinson.
- Si el deterioro cognitivo aparece antes, al mismo tiempo o dentro del primer año del inicio de los síntomas motores → Demencia con cuerpos de Lewy.
20.3.5 Tratamiento y cuidados
El tratamiento de la demencia por CL está todavía en estudios en los que se están probando, esencialmente, los mismos tratamientos que en la enfermedad de Alzheimer.
La levodopa no parece ser útil para los síntomas de parkinsonismo.
Los cuidados irán dirigidos a mejorar la calidad de vida y a evitar accidentes. Son cuidados similares a los de la enfermedad de Alzheimer con la particularidad de la dificultad motora que presentan estos pacientes (caídas, atragantamientos, inestabilidad...).
Quizá esta fórmula (aproximada) ayude a recordar esta enfermedad:
Enfermedad por CL = enfermedad de Alzheimer + enfermedad de Parkinson + delirio
20.4 DEMENCIAS FRONTOTEMPORALES
20.4.1 Concepto y epidemiología
Estas demencias son un grupo de enfermedades neurodegenerativas que se caracterizan clínicamente por alteraciones prominentes del comportamiento y/o del lenguaje, y patológicamente por atrofia cerebral focal por lo general de los lóbulos frontales y/o temporales. Estas demencias son también con conocidas como Síndrome frontal cerebral o demencias del espectro frontotemporal. La enfermedad de Pick es una de sus formas histopatológicas clásicas.
Es poco frecuente, pero es la segunda causa de demencia en menores de 65 años.
Se desconoce la etiología, pero la mayoría de los pacientes tiene historia familiar de la enfermedad.
20.4.2 Clínica
Los factores que más destacan clínicamente son los siguientes:
- Cambio precoz en el comportamiento: suele ser un cuadro muy llamativo con desinhibición social y/o sexual, apatía, perseveración, hiperfagia, labilidad emocional, ausencia de empatía, verborrea... La anosognosia es frecuente (no ser consciente de estar enfermo).
- Afasia no fluente: anomia, trastorno de repetición, tartamudeo, habla vacilante...
- Evolución progresiva hacia la demencia semántica: no comprender las palabras, su significado. También se disminuye la producción del lenguaje.
Es importante destacar que la pérdida de memoria y la apraxia suelen aparecer cuando la enfermedad está avanzada. Como la demencia por cuerpos de Lewy, puede tener afectada la motoneurona con sus consecuencias motoras.
20.5 DEMENCIAS VASCULARES
20.5.1 Concepto y epidemiología
La demencia vascular es el tipo de demencia que aparece tras episodios de accidente cerebrovascular. Su origen puede ser trombótico, embólico o hemorrágico.
Es la 2.ª causa de demencia en la persona mayor, en torno al 20-25 %.
20.5.2 Etiología
La etiología será la misma que para los ictus, así que la aterosclerosis, la hipertensión arterial, el tabaquismo, las arritmias y las aneurismas, entre otros, pueden ser desencadenantes de una demencia vascular.
20.5.3 Clínica
La clínica de la demencia vascular depende del tipo de trastorno circulatorio ocurrido y de la zona afectada, de modo que puede estar alterado el lenguaje, el pensamiento, la memoria, la movilidad, etc.
La evolución es menos previsible que en otras demencias. Puede decirse que, en general, son estables tras el ictus. Sin embargo, cualquier nuevo episodio cerebrovascular aumentará el grado de demencia, como ocurre, por ejemplo, en los multiinfartos cerebrales.
Entre la amplia variedad de cuadros, destacan estos:
- Demencia multiinfarto causada por la repetición de infartos cerebrales. Cada episodio aumenta el deterioro cognitivo y el deterioro general. Este tipo es el que más se aproxima a una demencia clásica como la enfermedad de Alzheimer en cuanto a la evolución.
- Demencia por infarto estratégico: se da en una localización que afecta a varias funciones cognitivas.
- Demencia postictus: instauración muy brusca. Normalmente, se acompañan de hemiparesia, disfagia y disartria.
- Demencia mixta: la persona padece la enfermedad de Alzheimer y demencia vascular.
- Demencia por lesiones hemorrágicas.
20.5.4 Tratamiento y cuidados
La prevención de nuevos episodios, la rehabilitación y los cuidados generales de la demencia son el modo de tratar estas demencias.
Como resumen, en el gráfico 6 se puede ver la distribución de las demencias más frecuentes.
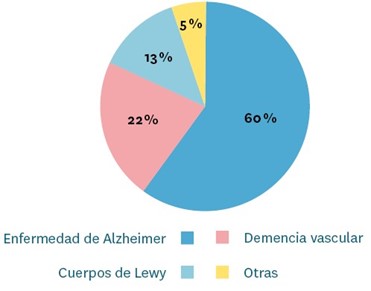
Gráfico 1. Tipos de demencias más frecuentes
20.6 Demencias rápidamente progresivas
20.6.1 Definición y concepto
Se denomina demencia rápidamente progresiva (DRP) a un síndrome caracterizado por un deterioro cognitivo y funcional que progresa en semanas o meses, siempre en menos de 1 o 2 años desde el inicio de los síntomas hasta una demencia clara con pérdida de autonomía en las actividades básicas de la vida diaria.
Este patrón se diferencia de las demencias típicas (por ejemplo, enfermedad de Alzheimer) donde la progresión suele medirse en años.
Desde el punto de vista clínico, las DRP son una urgencia neurológica, ya que algunas causas son potencialmente reversibles si se diagnostican y tratan precozmente.
20.6.2 Etiología de las DRP
Las DRP son un síndrome con múltiples causas posibles. Las principales etiologías son:
- Enfermedades neurodegenerativas: priónicas y no priónicas.
- Causas tóxico metabólicas: alteraciones hidroelectrolíticas, hepáticas, renales, endocrinas, déficits vitamínicos, fármacos.
- Causas infecciosas: encefalitis virales, VIH, sífilis, neurosífilis, enfermedad de Whipple, etc.
- Causas vasculares: infartos múltiples, vasculitis del SNC y fístulas arteriovenosas durales entre otras.
- Causas autoinmunes/paraneoplásicas.
- Causas neoplásicas: linfoma del SNC, carcinomatosis meníngea, tumores infiltrantes.
- Causas iatrogénicas: instrumentos neuroquirúrgicos contaminados, hormonas hipofisarias de origen humano, trasplantes de córnea o duramadre.
En la práctica clínica, y en muchas preguntas tipo test, la causa prototípica y más frecuente de DRP son las enfermedades priónicas.
20.6.3 Enfermedades priónicas: conceptos básicos
Las enfermedades priónicas son enfermedades neurode-generativas producidas por la acumulación de una forma anómala de la proteína prión (PrPSc) a partir de la proteína prión celular normal (PrPc).
- La PrPc es una proteína normal de membrana, expresada sobre todo en neuronas.
- La forma patológica PrPSc se pliega de manera anómala, es más resistente a la proteólisis y es capaz de inducir el mal plegamiento de nuevas moléculas de PrPc, actuando como “plantilla” autorreplicativa.
- Características generales de las enfermedades priónicas:
o Curso progresivo y habitualmente rápido.
o Afectación del sistema nervioso central con degeneración espongiforme: Al observar el cerebro al microscopio en estas enfermedades, se ven múltiples vacuolas (agujeritos) dentro de las neuronas y en la sustancia gris, de modo que el tejido adquiere un aspecto de esponja.
o Letales en todos los casos, sin tratamiento curativo disponible.
o Incidencia global alrededor de 1–2 casos por millón de habitantes/año.
o Consideradas enfermedades de declaración obligatoria en España y la UE.
20.6.3.1 Clasificación según la forma de aparición
Las enfermedades priónicas humanas pueden ser:
- Esporádicas (80–95%):
o No se identifica causa clara.
o Se atribuyen a una conversión espontánea de PrPc a PrPSc.
o Ejemplo: enfermedad de Creutzfeldt Jakob esporádica. - Genéticas o familiares (10–15%):
o Asociadas a mutaciones en el gen PRNP: es el gen humano que codifica la proteína priónica celular normal.
o Ejemplos: enfermedad de Creutzfeldt Jakob genética y el insomnio familiar fatal.
- Adquiridas (<1%):
o Por exposición a material contaminado con priones.
o Transmisión iatrogénica.
o Transmisión alimentaria: consumo de carne o derivados contaminados (encefalopatía espongiforme bovina).
20.6.4 Enfermedad de Creutzfeldt Jakob
La enfermedad de Creutzfeldt Jakob (ECJ) es la enfermedad priónica humana más frecuente y la causa prototípica de DRP.
20.6.4.1 Epidemiología
- En España: aproximadamente 1,1 casos/millón/año.
- Edad típica de presentación en la forma esporádica: 70–79 años.
- Supervivencia media: alrededor de 6 meses desde el inicio de los síntomas.
- En torno al 90% de los pacientes fallece antes del año.
20.6.4.2 Formas clínicas
- ECJ esporádica:
Es la forma más frecuente (aprox. 85–90%).
- ECJ genética o familiar:
Asociada a mutaciones en PRNP, con historia familiar en algunos casos.
- ECJ adquirida:
Incluye formas iatrogénicas y la variante de encefalopatía espongiforme bovina.
20.6.4.3 Clínica de la ECJ esporádica
La presentación típica incluye:
- Demencia subaguda con deterioro cognitivo rápido: memoria, funciones ejecutivas, conducta.
Síntomas motores:
- Ataxia: inestabilidad de la marcha, dificultad de coordinación.
- Mioclonías: sacudidas musculares breves, especialmente presentes con estímulos.
- Hipertonía y rigidez muscular.
Síntomas prodrómicos frecuentes: cefalea, apatía, depresión, vértigo, cambios conductuales.
Progresión a déficits piramidales, extrapiramidales, visuales cerebelosos y mayor deterioro cognitivo.
Fase final con mutismo acinético, encamamiento y fallo neurológico global.
20.6.4.4 Exploraciones complementarias
- Resonancia magnética cerebral: herramienta clave para diagnóstico de la enfermedad y como diagnóstico diferencial. Hallazgos:
o Hiperintensidad en secuencias de difusión (DWI) y FLAIR.
o Afectación característica de la corteza cerebral, dando el signo de "corteza en cinta" (cortex ribboning o encintado cortical): en la RM con difusión/FLAIR se observa una hiperintensidad lineal brillante que sigue las circunvoluciones de la corteza cerebral, como si se hubiera dibujado con un rotulador fluorescente sobre la superficie cortical.
o Afectación de los núcleos basales: caudado y putamen - Electroencefalograma (EEG).
- Estudio de líquido cefalorraquídeo (LCR).
La combinación de clínica típica, RM compatible y positividad de biomarcadores en LCR es muy útil para el diagnóstico probable de ECJ en vida.
20.6.4.5 Criterios diagnósticos
Sin entrar en todos los detalles, el diagnóstico de ECJ probable se basa en:
- Demencia rápidamente progresiva.
- Uno o más rasgos típicos (mioclonías, alteraciones visuales o cerebelosas, signos piramidales/extrapiramidales, mutismo acinético).
- Y al menos una de las siguientes pruebas positivas:
o EEG típico con complejos periódicos.
o Positividad de proteína 14-3-3 en LCR (con enfermedad de duración
o RM compatible
o Positividad del ensayo RT QuIC (biomarcador) en LCR.
- Ausencia de otra causa que explique mejor el cuadro.
20.6.4.6 Pronóstico y tratamiento
- La ECJ es una enfermedad siempre letal, con supervivencia media aproximada de 6 meses y rara vez superior a 1 año.
- No existe tratamiento curativo ni fármacos que hayan demostrado modificar la progresión de la enfermedad.
El manejo se centra en:
- Cuidados paliativos y de confort.
- Control sintomático (mioclonías, agitación, dolor).
- Apoyo a la familia y planificación anticipada de cuidados.
- Medidas de bioseguridad en procedimientos de riesgo (instrumental neuroquirúrgico, LCR, etc.).
20.6.5 Variante de la enfermedad de Creutzfeldt Jakob: encefalopatía espongiforme bovina
La variante de ECJ (vECJ) es una forma adquirida ligada a la exposición al prion bovino de la encefalopatía espongiforme bovina (EBB), también conocido como “el mal de las vacas locas”.
20.6.5.1 Epidemiología
- Descrita por primera vez en Reino Unido en 1996.
- Afecta a personas más jóvenes que la ECJ esporádica, a menudo menores de 40 años.
- Se han asociado los casos al consumo de productos bovinos contaminados con EEB.
20.6.5.2 Clínica característica
En la vECJ, el inicio suele diferir de la forma clásica:
- Debut con síntomas psiquiátricos prominentes: ansiedad, depresión, cambios conductuales.
- Parestesias dolorosas en extremidades.
- Evolución posterior con:
o Alucinaciones y delirios.
o Conducta agresiva o desinhibida.
o Ataxia, mioclonías, corea y distonía.
Presenta un curso progresivo igualmente letal, aunque la supervivencia puede ser algo más prolongada que en la forma esporádica en algunos casos. Su diagnóstico y manejo es el mismo que en las otras formas clínicas. La confirmación definitiva es post-mortem.
20.6.5.3 Diagnóstico y manejo
El diagnóstico se basa en:
- Historia de exposición posible a productos de riesgo (EEB).
- Clínica típica: inicio psiquiátrico en adultos jóvenes.
- Neuroimagen y pruebas de LCR/RT QuIC.
- Confirmación definitiva post mortem.
El manejo es también fundamentalmente paliativo, con las mismas consideraciones de bioseguridad.
20.6.6 Rol de enfermería en las DRP y ECJ
El papel de enfermería es fundamental tanto en el proceso diagnóstico como en los cuidados continuados.
20.6.6.1 Valoración y cuidados básicos
- Valoración integral del paciente con escalas funcionales y cognitivas.
- Prevención de úlceras por presión, contracturas y complicaciones de inmovilidad.
- Manejo de la disfagia (adaptación de textura, riesgo de aspiración).
- Control de síntomas: dolor, agitación, ansiedad, insomnio, mioclonías.
- Apoyo emocional al paciente y familia, educación sobre la evolución esperable.
20.6.6.2 Bioseguridad y prevención de riesgos
Aunque el contagio en el entorno habitual es extremadamente bajo, en contextos sanitarios se deben extremar las medidas de bioseguridad con materiales de riesgo:
- Especial precaución con tejido nervioso, LCR y material quirúrgico utilizado en SNC y ojos.
- Uso de protocolos específicos de descontaminación de priones para instrumental
- Registro y comunicación adecuada al equipo de medicina preventiva y salud pública cuando se confirma una prionopatía ya que es una enfermedad de declaración obligatoria.
20.7 ENFERMEDAD DE PARKINSON
20.7.1 Concepto y epidemiología
La enfermedad de Parkinson (EP) es una enfermedad degenerativa causada por una pérdida de las neuronas dopaminérgicas en la sustancia negra del mesencéfalo. Este proceso conlleva una disminución de la secreción de dopamina provocando los signos principales de la enfermedad: temblor de reposo, bradicinesia, rigidez e inestabilidad postural. Se trata de un proceso crónico, progresivo y degenerativo que se enmarca dentro de los trastornos del movimiento.
Aunque hay multitud de enfermedades y procesos que pueden producir parkinsonismo (signos similares a la EP), la enfermedad de Parkinson idiopática o Parkinson primario produce el 85 % de ellos. En este capítulo, cuando aparece enfermedad de Parkinson se hace referencia a la idiopática o Parkinson primario.
Es la 2.ª enfermedad neurodegenerativa más frecuente tras la enfermedad de Alzheimer (no es la 2.ª causa de demencia, pues ese puesto lo ocupa la demencia vascular).
Se estima que el 1,5 % de los mayores de 65 años y el 10 % de los mayores de 80 años pueden padecer la EP.
20.7.1 Etiología
Se desconoce el origen de la enfermedad, pero se especula con la propia vejez, la predisposición genética (solo el 10% de los casos) y la exposición a algunas sustancias (pesticidas, herbicidas...).
20.7.3 Clínica
Los signos aparecen tras unos 5-10 años de periodo preclínico, ya que el inicio de los síntomas ocurre cuando se han perdido el 60% de las células dopaminérgicas y el nivel de dopamina se ha reducido un 80%. Al principio, la clínica es pequeña, pero, a medida que se van perdiendo neuronas dopaminérgicas y va aumentado la zona hipofuncional, los signos van siendo más potentes.
Estos son los principales signos:
- Temblor de reposo:suele ser el primer signo que aparece, en el inicio en una sola extremidad. Es un temblor de una frecuencia de 4-6 Hz en los dedos de la mano como “de contar monedas” o de “rodar la píldora”. Es el signo menos invalidante. Afecta principalmente a brazos y piernas, y en menor medida a labios, lengua y mentón. Por la noche el temblor se reduce e incluso desaparece.
- Rigidez:hipertonía muscular con resistencia aumentada y sostenida. Cuando se combina con el temblor, el movimiento de la articulación es en “rueda dentada”. Rigidez dolorosa y molesta.
- Inestabilidad postural:disminuido el reflejo de corregir la postura, se explora fácilmente con la prueba del empujón.
- Bradicinesia: Es la lentitud de movimientos, incluso la pérdida de movimientos automáticos o retraso en su ejecución. Se manifiestan los siguientes signos:
o Dificultad para realizar tareas de psicomotricidad fina (abrocharse los botones, atarse los zapatos). También, dificultad para levantarse, sentarse, dar giros...
o Hipomimia: falta de expresión facial. “Aspecto de máscara” o “cara de Póquer”.
o Micrografía: letra pequeña en la escritura.
o Hipofonía: voz más baja.
o Bloqueo motor o congelación parkinsoniana: parada súbita mientras se deambula, al girar o al pasar por obstáculos o sitios estrechos. Sensación de que los pies se quedan pegados al suelo.
Otros signos importantes:
- Disfunción del sistema nervioso autónomo:sialorrea, estreñimiento, seborrea, incontinencia urinaria.
- Trastornos de la marcha:marcha típica conocida como marcha festinante: postura en flexión, dificultad al iniciar la marcha, arrastre de pies, pasos cortos, ausencia de braceo, giros en bloque.
- Dolor y parestesias.
- Disfagia:el 90 % de los pacientes en fases avanzadas. También puede aparecer en fases más tempranas, pero es menos frecuente.
- Demencia:la desarrollan casi la mitad de los pacientes (sobre todo los más mayores) en fases avanzadas de la enfermedad.
- Emocionales:depresión y ansiedad.
- Caídas por la marcha festinante, la inestabilidad postural y la rigidez.
- Otros: hiposmia, trastornos del sueño...
Según la clasificación de Hoehn y Yahr, los estadios y signos de la enfermedad se distribuyen de la siguiente manera:
Clasificación de Hoehn y Yahr
- No hay signos de enfermedad
- Afectación exclusivamente unilateral
- Afectación bilateral, sin alteración del equilibrio (prueba del empujón normal)
- Afectación moderada; cierta inestabilidad postural, pero físicamente independiente.
- Incapacidad grave, aún capaz de caminar o de permanecer en pie sin ayuda
- Permanece en silla o en cama.
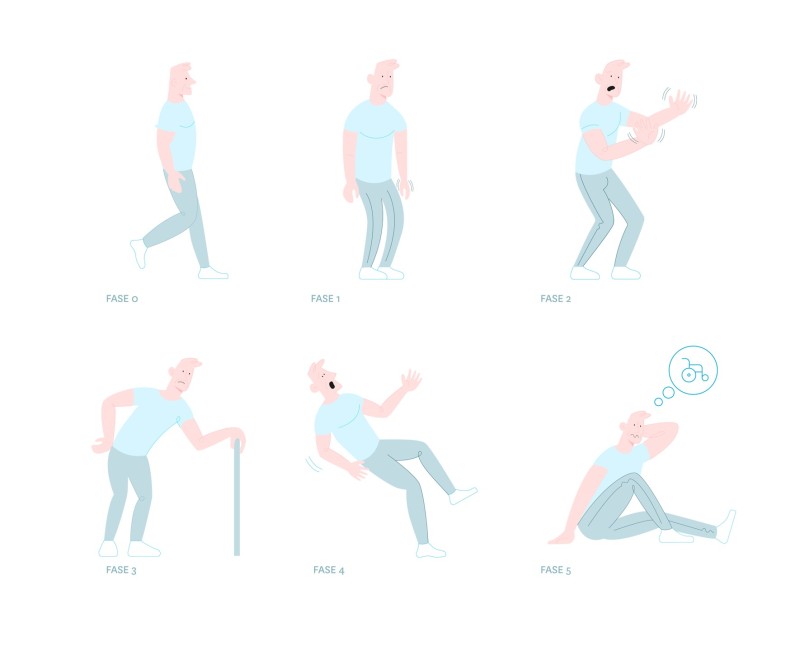
Imagen 13. Clasificación de los estadios de la enfermedad de Parkinson según Hoenh y Yarh
La escala MDS-UPDRS (Movement Disorder Society – Unified Parkinson’s Disease Rating Scale) es la más utilizada actualmente para valorar la enfermedad de Parkinson. Evalúa:
- Síntomas motores y no motores
- Impacto en las actividades de la vida diaria
- Percepción del propio paciente
Es útil tanto para el seguimiento clínico como para la investigación, y permite valorar la eficacia del tratamiento. A diferencia de Hoehn y Yahr, que clasifica de forma global, la MDS-UPDRS ofrece un detalle cuantitativo y permite hacer un seguimiento preciso de la evolución del paciente.
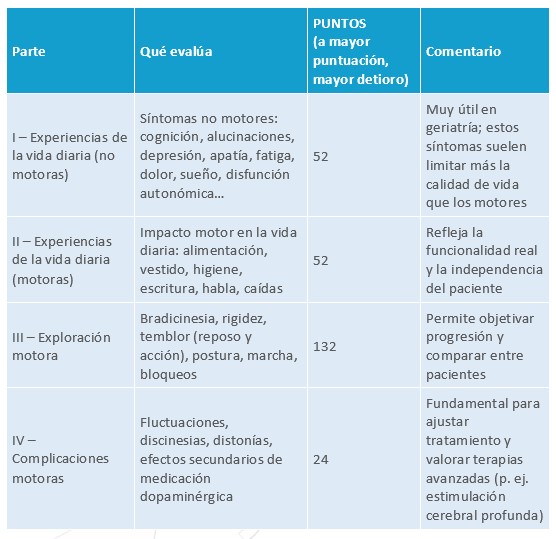
Interpretación práctica:
- Se suman los ítems de cada parte para obtener una puntuación total de 0 a 260.
- Puntuaciones más altas reflejan peor estado del paciente, tanto motor como no motor.
- En la práctica clínica se utiliza más para comparar evolución de un paciente en el tiempo o evaluar eficacia de tratamientos, no para clasificar “leve/moderado/severo” de manera absoluta.
20.7.4 Diagnóstico
El diagnóstico de la EP es fundamentalmente clínico. Se basa en los síntomas descritos, en su ritmo de aparición y descartando el resto de enfermedades que puedan causar el síndrome parkinsoniano.
Además de los signos y síntomas, también se buscan antecedentes familiares, se descarta otro tipo de parkinsonismo...
También se usa la respuesta a levodopa para orientar el diagnóstico puesto que el 70-100 % de los pacientes con EP idiopática responden adecuadamente al tratamiento.
20.7.5 Tratamiento y cuidados
El tratamiento farmacológico:
- Levodopa: el más eficaz para la bradicinesia y la rigidez. Habitualmente se administra por vía oral en comprimidos de liberación rápida o retardada, también existen suspensiones orales y administración inhalada, esta última vía es útil para el alivio rápido de los síntomas motores en episodios off. La vida breve de este medicamento produce efectos wearing-off (aumento de la sintomatología cuando va acabando el efecto) y, cuando la enfermedad avanza, pueden aparecer efectos on-off (aumento y disminución de la sintomatología de manera impredecible entre dosis, los efectos on-off suelen aparecer tras 5-7 años de tratamiento con levodopa). Estos efectos son más visibles en la bradicinesia y el temblor. En algunos pacientes puede llegar a utilizarse una opción de 2.ª línea como la irrigación intestinal continua de levodopa/carbidopa.
- Agonistas dopaminérgicos: como monoterapia en fases iniciales para retrasar el uso de la levodopa o como adyuvante de la propia levodopa. Un ejemplo es la rotigotina que se administra en parches transdérmicos de 24 horas de duración. Efectivo para el temblor. La apomorfina se usa por vía subcutánea exclusivamente y es un tratamiento de 2.ª línea. Se usan infusores para la perfusión continua o plumas precargadas para la administración intermitente. La apomorfina se utiliza para el control de las fluctuaciones motoras severas en Parkinson avanzado, hace su efecto en unos 20 minutos y desaparece en 2 horas.
- IMAO: eficacia sobre los síntomas motores.
- Anticolinérgicos: efectivos para el temblor, la sialorrea y la distonía. Aumentan el estreñimiento y la retención urinaria. Es el que menos evidencia tiene y mayores efectos adversos presenta.
- Antidepresivos, ansiolíticos, analgésicos, laxantes...
- Antiheméticos: en caso de problemas gástricos o de vómitos derivados del propio Parkinson o de sus tratamientos, se recomienda la domperidona, quedando contraindicada la metoclopramida porque empeora los síntomas motores.
- En caso de demencia puede usarse los IACEs, pero si aumentan mucho la sialorrea pueden suspenderse.
Tratamiento no farmacológico:
- Fisioterapia.
- Logopedia: para la hipofonía y la dificultad de deglución.
- Terapia ocupacional.
- Quirúrgico: está cobrando importancia la estimulación cerebral profunda porque mejora la sintomatología. Prácticamente ha sustituido a las técnicas ablativas (talamotomía, subtalamotomía...). Su usan cuando el tratamiento farmacológico no es efectivo o es insuficiente.
Cuidados de enfermería: como personal de Enfermería, se informa al paciente y a la familia sobre la enfermedad y su evolución, así como de los cuidados oportunos. De la misma manera, se motiva para que la persona aumente y mantenga su capacidad funcional. A continuación se listan los principales cuidados:
- Alimentación: adecuar la dieta a semisólida por la dificultad de deglutir; además pueden espesarse los líquidos. Indicar que no hable mientras se come para reducir el riesgo de aspiración. Si se distribuye la ingesta de proteínas durante el día aumentando su presencia por la noche, se disminuyen los efectos on-off de la levodopa.
- Higiene: diaria. Estos pacientes tienen mucha sudoración por la alteración del sistema nervioso autónomo. Fomentar la autonomía adaptando el espacio y los utensilios. Evitar lociones oleosas porque existe una producción seborreica excesiva.
- Ropa: adaptar las prendas a la funcionalidad o sustituir por otras que faciliten el vestirse y desvestirse.
- Evitar las altas temperaturas porque algunos fármacos antiparkinsonianos pueden impedir la pérdida de calor de la persona al limitar su sudoración.
- Mobiliario: muebles adaptados que aumenten la autonomía.
- Cuidados encaminados a evitar caídas.
- Cuidados del estreñimiento, la incontinencia o la retención urinaria.
- Fomentar el ejercicio y unos hábitos de vida saludables.
- Informar del tratamiento farmacológico y ayudar a distribuir las dosis para evitar los efectos on-off.
- Vigilar el estado emocional e informar de los recursos sociales disponibles.
- Recomendar asociaciones, federaciones, grupos y demás recursos sociales. La mayoría de las asociaciones han adoptado el tulipán como símbolo.
BIBLIOGRAFÍA
- Del Castillo Martín F, Baquero Artigao F, de la Calle Cabrera T, et al. Documento de consenso sobre etiología, diagnóstico y tratamiento de la otitis media aguda. Anales de Pediatría. 2012;77(5),345.e1-345.e8. doi: 10.1016/j.anpedi.2012.05.02
- Ministerio de Sanidad, Servicios Sociales e Igualdad. [Internet] Guías de Práctica Clínica en el SNS. Guía de Práctica Clínica sobre Glaucoma de Ángulo Abierto;2017. [citado 21 de marzo de 2021]. Recuperado a partir de: https://portal.guiasalud.es/wp-content/ uploads/2018/12/GPC_568_Glaucoma_AQUAS_compl.pdf
- Fuster Peiró MA, García Velasco MP, Gómez Vela ML, Herrera Barcia T, Lendínez Mesa A, Monleón Just M, Sánchez Martínez N. Guía de recomendaciones prácticas en Enfermería. Dolor neuropático periférico. Fontán Vinagre G, Guerrero Menéndez R, Monleón Just M (coordinadores). Madrid: Instituto de Investigación del Consejo General de Enfermería; 2024.
https://www.consejogeneralenfermeria.org/profesion/guias-clinicas/send/160-guias-clinicas/2870-dolor-neuropatico-periferico?utm_source=chatgpt.com - Latorre G, González García N, García Ull J, González Oria C, Porta Etessam J, Molina FJ, et al. Diagnóstico y tratamiento de la neuralgia del trigémino: documento de consenso del Grupo de Estudio de Cefaleas de la Sociedad Española de Neurología. Neurología. 2023 Jun;38(1):S37–52.
- Lassaletta L, Morales-Puebla JM, Altuna X, Arbizu Á, Arístegui M, Batuecas Á, et al. Parálisis facial: Guía de práctica clínica de la Sociedad Española de Otorrinolaringología y Cirugía de Cabeza y Cuello. Acta Otorrinolaringológica Española. 1010;71(2):99-118.
- Garzón-Giraldo MLD, Montoya-Arenas DA, Carvajal-Castrillón J. Perfil clínico y neuropsicológico: enfermedad de Parkinson/enfermedad por cuerpos de Lewy. Rev CES Med. 2015;29(2):255-270.
- Millán Calenti JC. Gerontología y Geriatría: valoración e intervención. Madrid: Editorial Médica Panamericana; 2010.
- Ministerio de Sanidad, Consumo y Bienestar Social. Sanidad [Internet]. Plan Integral de Alzheimer y otras Demencias (2019-2023); 2019. [citado 21 de marzo de 2021]. Recuperado a partir de: https://www.mscbs.gob.es/profesionales/saludPublica/docs/Plan_Integral_Alhzeimer_Octubre_2019.pdf
- Ministerio de Sanidad, Política Social e Igualdad [Internet]. Guías de práctica clínica en el SNS. Guía de Práctica Clínica sobre la Atención Integral a las Personas con Enfermedad de Alzheimer y otras Demencias; 2011. [citado 21 de marzo de 2021]. Recuperado a partir de: https://portal.guiasalud.es/gpc/guia-de-practica-clinica-sobre-la-atencion-integral-a-las-personas-con-enfermedad-de-alzheimer-y-otras-demencias/
- Olivares Romero J, Escamilla Sevilla F, editores. Recomendaciones de práctica clínica en la Enfermedad de Parkinson.
- [Internet] Sociedad Andaluza de Neurología. Grupo Andaluz de Trastornos del Movimiento. Barcelona: Editorial Glosa; 2017. [citado 21 de marzo de 2021]. Recuperado a partir de: https://portal.guiasalud.es/gpc/enfermedad-parkinson/
- Sociedad Española de Geriatría y Gerontología [Internet]. Tratado de Geriatría para residentes. [citado 21 de marzo de 2021]. Recuperado a partir de: https://www.segg.es/tratadogeriatria/main.html
- Sociedad Española de Geriatría y Gerontología [Internet].
- Manual del residente en Geriatría; 2011. [citado 21 de marzo de 2021]. Recuperado a partir de: https://www.segg.es/media/descargas/Acreditacion%20de%20Calidad%20SEGG/CentrosDia/ManualResidenteGeriatria-2.pdf