3.1 INTRODUCCIÓN
El tratamiento del cáncer infantil ha mejorado de forma espectacular en los últimos 25 años, con un aumento de las tasas globales de curación entre el 20-30% a finales de los 70, hasta por encima del 75% en la actualidad. Sin embargo, no se puede caer en la complacencia pensando que lo más importante está hecho.
El cáncer infantil es un grave problema en los países desarrollados, aunque a veces no se le presta la atención que merece, porque queda oculta en la abundancia de los cánceres propios de la edad adulta y la vejez. A pesar de los grandes avances terapéuticos, el cáncer es la primera causa de muerte por enfermedad en la infancia y la adolescencia, a partir del primer año de vida. El Registro Nacional de Tumores Infantiles de España (RETI-SEHOP) recopila los datos sobre el cáncer infantil en España desde el año 1979.
Ante la práctica imposibilidad de prevención primaria, la lucha contra el cáncer infantil reside principalmente en el máximo uso de las posibilidades diagnósticas y terapéuticas que ofrece la oncología actual. Con la rapidez de los avances asistenciales, cobra cada vez mayor importancia la comparación de los resultados de supervivencia entre países, como medida diferencial y para la monitorización del acceso oportuno de los niños que enferman de cáncer en distintas poblaciones a la asistencia apropiada.
En Europa, se realiza una importante tarea, basada en métodos epidemiológicos rigurosos, dedicada a la comparación de los resultados de supervivencia de los enfermos de cáncer entre países y regiones, como forma de evaluar y detectar posibles déficits asistenciales; así como, para estudiar las causas de las diferencias que se observan en los resultados entre distintos países.
En relación con las causas del cáncer infantil, los escasos factores de riesgo confirmados explican muy pocos casos. El origen es multifactorial, incluyendo una variedad de factores ambientales (físicos, químicos y biológicos) y genéticos, donde la susceptibilidad individual sería importante y la interacción entre factores genéticos y ambientales todavía es ampliamente desconocida. Algunas alteraciones genéticas predisponentes podrían tener origen medioambiental y algunos factores podrían actuar antes del nacimiento o de la concepción. Hasta hoy, los estudios epidemiológicos no han podido identificar riesgos medioambientales específicos. En general, los factores de riesgo sospechosos son hipótesis que requieren confirmación.
3.2 INCIDENCIA Y SUPERVIVENCIA DEL CÁNCER INFANTIl
En el año 2023, se registraron un total de 993 casos nuevos de niños con tumores entre 0 y 14 años residentes en España, según el Registro Español de Tumores Infantiles (RETI-SEHOP) de estadísticas desde 1980 a 2023. Además, incluido en este registro, se diagnosticaron 150 casos nuevos de adolescentes ente 15 y 19 años durante el 2023. Aunque poco frecuente, el cáncer infantil tiene gran impacto sobre las familias y es un importante problema de salud. Los resultados asistenciales han progresado mucho, pero en los países desarrollados es la primera causa de muerte por enfermedad en la infancia. Los pacientes curados pueden tener secuelas de por vida. El cáncer infantil no puede ser prevenido, la lucha reside en el diagnóstico rápido y el inicio del tratamiento.
El propósito del Registro Español de Tumores Infantiles (RETI-SEHOP) es investigar el cáncer infantil y su asistencia en España, su incidencia y supervivencia, y contribuir al conocimiento de sus causas.
Las posibilidades de sobrevivir a un cáncer en nuestro país, en menores de 14 años, son del 82% a los 5 años, de acuerdo con los últimos datos del Registro Español de Tumores Infantiles (RETI-SEHOP 2023), subiendo un 5% respecto a los últimos 15 años.
Por tanto, no se van a curar al 18-20% de niños y adolescentes. Por esto, es importante contar con equipos de apoyo, tanto Unidades de Cuidados Paliativos Pediátricos como Hospitalización Domiciliaria, para garantizar la mejor atención y calidad de vida del paciente y su familia en la última fase de su enfermedad.
Los análisis internacionales, como el CONCORD-3, muestran cifras superiores al 85% en Canadá, Chipre, Bélgica, Dinamarca, Finlandia y Australia, y advierte de la necesidad de mejorar la supervivencia de estos pacientes en España, sobre todo en leucemias y tumores cerebrales.
Asimismo, destacar que más del 70% de los supervivientes de un cáncer en la edad pediátrica presentarán secuelas a largo plazo derivadas del tratamiento, que pueden condicionar su integración académica, laboral, social y afectiva.
3.3 VALORACIÓN DE ENFERMERÍA
3.3.1 Semiología en oncología
Para entender la información sobre la extensión de la enfermedad se emplea la guía de la OMS que trata sobre TNM Esencial donde se definen unas instrucciones generales.
- Principios TNM Esencial:
El TNM esencial está conformado por tres elementos que en su conjunto resumen la extensión del cáncer en el paciente. Estos elementos son:
- T: extensión de la invasión y/o tamaño del tumor
- N: presencia o ausencia de ganglios (nodos) regionales/ compromiso regional
- M: presencia o ausencia de metástasis distantes
- Clasificación de los elementos:

Metástasis a distancia (M) significa que el tumor original (primario) se ha diseminado a órganos distantes o ganglios linfáticos distantes (no regionales). Se basa en la mejor información disponible, ya sea clínica, hallazgos en cirugía, imágenes o patología.

El compromiso de los ganglios linfáticos implica que el tumor se ha diseminado a través del sistema linfático y que se encuentran células cancerosas en los ganglios linfáticos que drenan el órgano específico. En este caso, N se basa en los datos más específicos disponibles para confirmar la presencia o ausencia de compromiso del nodo regional y generalmente se clasifica a partir del informe de patología. Un nódulo "agrandado" o "palpable" no constituye compromiso si se basa únicamente en esas palabras.

Por tanto, T se basa en los datos más específicos disponibles para confirmar el grado de invasión dentro / a través del órgano involucrado y / o el tamaño del tumor primario (según el tipo del cáncer). Generalmente se codifica a partir del informe de patología y clasifica ampliamente la extensión como avanzada o limitada. En este caso, T puede codificarse a partir del expediente clínico (endoscopia, radiografías, palpación) en ausencia de confirmación patológica.
- Asignación de Estadio en TNM Esencial:
Una vez que los elementos del TNM Esencial se han codificado, se pueden combinar en grupos de estadio que van del I a IV, de acuerdo con la gravedad de la enfermedad. Los grupos de estadio fueron diseñados para agrupar pacientes con cáncer en categorías de previsión similares.
- Estadio IV para cánceres con metástasis a distancia.
- Estadios III y II para cánceres con mayor o menor grado de compromiso ganglionar.
- Estadio I se suele asignar a cánceres con un compromiso limitado.
Las reglas para la combinación de los elementos del TNM Esencial en los grupos de estadio (IV-I) se proporcionan de forma específica para cada tipo de cáncer.
3.3.2 Signos y síntomas frecuentes del cáncer infantil:
Es posible que el cáncer infantil se presente con signos y síntomas que son compartidos por otras enfermedades infantiles. Aunque un solo hallazgo aislado no siempre requiere evaluación para el cáncer, una combinación de múltiples hallazgos (por ejemplo: pérdida de peso, dolor óseo y linfadenopatía; hematomas fáciles, recuentos sanguíneos anormales y hepatoesplenomegalia) es preocupante y, por lo general, justifica la evaluación de la neoplasia maligna. Además, ciertos hallazgos son preocupantes incluso como hallazgos aislados (por ejemplo: masas abdominales o mediastínicas, cefaleas asociadas con vómitos por la mañana, blastocitos en el frotis de sangre periférica) y requieren una evaluación y consulta inmediatas.
Se entiende por síntomas constitucionales inespecíficos: fiebre, pérdida de peso y fatiga, que son hallazgos comunes en los niños con cáncer.
- Fiebre:
La fiebre es un signo común en los niños y rara vez se puede atribuir a una neoplasia maligna.
Cuándo sospechar de una neoplasia maligna: se debe plantear la sospecha de causas no infecciosas cuando la enfermedad febril no sigue el curso habitual o no responde a una terapia aparentemente adecuada. Sin embargo, incluso cuando la fiebre es prolongada, la infección sigue siendo la causa más común. En informes publicados de niños con fiebre prolongada, se encontró que una pequeña minoría (aproximadamente el 5%) tenía una neoplasia maligna, más comúnmente leucemia y linfoma.
En ausencia de infección, la fiebre persistente o recurrente puede reflejar una neoplasia oculta, como un linfoma. En raras ocasiones, puede ser secundaria a necrosis relacionada con el tumor, como en el caso del neuroblastoma o el tumor de Wilms. La fiebre persistente también puede ser el único signo en niños con leucemia, sarcoma de Ewing e histiocitosis de células de Langerhans. Más de la mitad de los niños con leucemia tienen fiebre en el momento de la presentación.
Un examen físico completo puede revelar hallazgos preocupantes adicionales, como linfadenopatías o hepatoesplenomegalia.
- Pérdida de peso:
La mayoría de los niños que presentan una pérdida de peso aislada tienen etiologías no malignas (por ejemplo: deshidratación, infección, desnutrición). Sin embargo, los pacientes con pérdida de peso continua e involuntaria en el contexto de anemia, palidez, hematomas, dolores corporales, linfadenopatía, hepatoesplenomegalia, fiebres inexplicables o fatiga pueden tener una neoplasia maligna subyacente.
- Fatiga y palidez:
La disminución de la energía, la fatiga y la palidez pueden ocurrir en una amplia gama de enfermedades. La palidez se debe con mayor frecuencia a la anemia, pero también puede ocurrir en afecciones no hematológicas como infecciones crónicas, trastornos reumatológicos, insuficiencia cardíaca, arritmia o trastornos metabólicos. La fatiga y la palidez se deben con mayor frecuencia a causas no malignas. Sin embargo, la preocupación por la malignidad puede surgir si hay otros hallazgos acompañantes preocupantes (petequias, hematomas, linfadenopatía, hepatoesplenomegalia, dolor óseo). Las pruebas de laboratorio iniciales en niños con palidez generalmente incluyen un hemograma completo con recuento diferencial y de reticulocitos. Esto puede ayudar a identificar si hay otros hallazgos hematológicos preocupantes (por ejemplo: otras líneas celulares afectadas, blastocitos periféricos), que deben impulsar la consideración de la derivación y el examen de la médula ósea.
- Cefalea:
El dolor de cabeza es otro síntoma común en la práctica pediátrica general. Los tumores intracraneales son una causa poco frecuente de dolor de cabeza en los niños, pero se deben considerar cuando los dolores de cabeza son persistentes o empeoran en intensidad, particularmente si se asocian con vómitos, cambios visuales, debilidad asimétrica o dificultades de coordinación. Los antecedentes de dolor de cabeza en un niño, particularmente en un niño menor de 10 años, se obtienen mejor con la opinión de los padres.
- Linfadenopatía:
La linfadenopatía es otro hallazgo común en los niños. El tamaño de los ganglios linfáticos normales en los niños varía ampliamente a medida que los niños están expuestos a nuevos virus y bacterias. La mayoría de los niños tienen pequeños ganglios linfáticos cervicales, axilares o inguinales palpables en algún momento de la infancia. El tamaño de un ganglio linfático que se considera anormal varía según la región de los ganglios linfáticos y la edad del niño. El riesgo de malignidad aumenta en los ganglios linfáticos mayor de 2 cm de diámetro, aunque la malignidad puede ocurrir en ganglios más pequeños.
La mayoría de los ganglios agrandados están relacionados con causas benignas como la infección. Sin embargo, la linfadenopatía puede ser un signo de presentación de leucemia, linfoma y neuroblastoma. Por el contrario, la linfadenopatía es poco frecuente (aunque se observa ocasionalmente) con tumores de tejidos blandos, huesos y células germinales.
El sitio de la adenopatía y la edad del niño pueden ayudar a reducir el rango de posibles diagnósticos. Los cánceres más comunes asociados con la linfadenopatía de cabeza y cuello son el neuroblastoma, el rabdomiosarcoma, el linfoma no Hodgkin y la leucemia en niños menores de 6 años, mientras que los linfomas (tanto Hodgkin como no Hodgkin) predominan en niños entre 7 y 13 años. La biopsia escisional de ganglios linfáticos puede estar indicada en pacientes con características preocupantes.
- Dolor óseo y articular: El dolor óseo y articular puede ser un síntoma de presentación de tumores que comprometen el hueso o la médula ósea, como son:
Tumores óseos malignos: el sarcoma de Ewing y el osteosarcoma. El dolor asociado con los tumores óseos primarios generalmente comienza como un dolor intermitente y aumenta en gravedad con el tiempo, aunque puede aumentar y disminuir. Puede ser palpable una masa asociada en el examen. Las fracturas patológicas ocurren en aproximadamente el 10-15 % de los casos de sarcoma de Ewing u osteosarcoma.
Leucemia aguda: el dolor óseo es un síntoma de presentación en aproximadamente el 20-30% de los niños con leucemia aguda, y otros síntomas musculoesqueléticos (incluido el dolor de cadera, dolor en las extremidades, dolor en las articulaciones y cojera) ocurren en aproximadamente el 60-70%. El dolor nocturno y el dolor óseo no articular se asocian más comúnmente con la leucemia, mientras que la rigidez matutina y la erupción cutánea ocurren más comúnmente en condiciones reumatológicas.
- Masas mediastínicas:
Los tumores mediastínicos pueden ser asintomáticos o pueden estar asociados con síntomas como tos, dificultad para respirar (a menudo posicional), ronquera, sibilancias o hinchazón facial o del cuello. Cuando los síntomas están presentes, generalmente son el resultado de la compresión extrínseca o la afectación de estructuras adyacentes, como el nervio laríngeo recurrente. Las masas mediastínicas a menudo se descubren incidentalmente en las radiografías de tórax obtenidas por otras razones.
- Masas abdominales:
Una masa abdominal palpable, que a menudo es detectada por un miembro de la familia o un proveedor de atención primaria, es uno de los signos de presentación más comunes de tumores sólidos malignos en niños. El síntoma de presentación puede ser dolor, vómitos, estreñimiento o, con menor frecuencia, obstrucción intestinal. Aunque algunas masas abdominales son benignas, todas requieren un examen temprano y completo.
Tumores intraabdominales comunes: el tumor de Wilms y el neuroblastoma son los tumores intraabdominales más comunes. Otros menos frecuentes son: linfoma, tumores hepáticos, tumores de ovario y sarcomas de tejidos blandos.
La edad del niño ayuda en el diagnóstico diferencial. El tumor de Wilms y el neuroblastoma ocurren con mayor frecuencia en bebés y niños pequeños, mientras que la afectación leucémica o linfomatosa del hígado, el bazo o los ganglios linfáticos retroperitoneales ocurre con mayor frecuencia en niños mayores.
- Síntomas de sangrado:
Cuando el sangrado es el signo inicial de cáncer infantil, generalmente se debe a una trombocitopenia, que, a su vez, suele ser causada por la afectación neoplásica de la médula ósea. Esto se manifiesta típicamente como sangrado cutáneo (como petequias y equimosis) y/o sangrado de la mucosa (epistaxis, sangrado gingival, sangrado bucal).
- Anomalías en el hemograma:
Los recuentos sanguíneos anormales son una característica común que se presenta en las neoplasias malignas infantiles. Estos hallazgos pueden observarse en un hemograma obtenido para la evaluación de otros hallazgos preocupantes (por ejemplo: palidez, petequias, linfadenopatía, dolor óseo) o en un hemograma obtenido por otras razones (por ejemplo: cribado de rutina o evaluación de enfermedad febril).
3.3.3 Urgencias oncológicas
En algunos casos, es necesaria la administración de terapia de urgencia en el momento de la presentación para estabilizar el estado del niño. Algunos ejemplos son:
- Masas mediastínicas que causan compresión de las vías respiratorias, cardíacas o de los vasos sanguíneos principales.
- Tumores cerebrales que causan presión intracraneal elevada.
- Hiperleucocitosis por leucemia aguda, que puede causar morbilidad neurológica, incluyendo hemorragia intracraneal o accidente cerebrovascular isquémico.
- Hiperuricemia, alteraciones electrolíticas (hiperpotasemia, hiperfosfatemia, hipocalcemia) y lesión renal aguda por síndrome de lisis tumoral. Por lo general, el síndrome de lisis tumoral ocurre después del inicio de la terapia citotóxica, en cambio, puede ocurrir antes del tratamiento en neoplasias malignas que tienen una tasa proliferativa alta, como algunas formas de linfoma (por ejemplo: linfoma de Burkitt, linfoma de células T) y leucemia aguda.
3.4 CLASIFICACIÓN DE LA PATOLOGÍA ONCOLÓGICA PEDIÁTRICA:
3.4.1 Tumores del Sistema Nervioso Central (SNC):
El sistema nervioso central (SNC) se divide en tres compartimentos principales: la médula espinal, la región infratentorial y la región supratentorial. La región infratentorial incluye el tronco cerebral y el cerebelo, mientras que la región supratentorial incluye: los hemisferios cerebrales, el tálamo, ganglios de la base, diencéfalo, tractos ópticos/región quiasmática y área hipotálamo-hipofisaria.
Los signos y síntomas derivados de estas neoplasias dependen de diversos factores, como la localización del tumor, la tasa de crecimiento del mismo y la edad del niño.
Los tumores del SNC son las neoplasias más frecuentes en la infancia tras las leucemias y, globalmente, constituyen la primera causa de muerte por cáncer en la infancia. Su incidencia se sitúa entre 1 y 3 por 100.000 menores de 19 años (5,4 por 100.000 cuando se incluyen los tumores benignos).
Los tumores supratentoriales son más frecuentes en niños menores de 3 años y en mayores de 10, mientras que los infratentoriales son más frecuentes entre los 4 y 10 años (45-60% de los tumores cerebrales de los niños se localizan en la fosa posterior). A nivel histológico, dentro de los tumores infratentoriales, los más frecuentes son: gliomas cerebelosos y troncoencefálicos, y meduloblastomas, seguidos de los ependimomas. A nivel supratentorial predominan los astrocitomas.
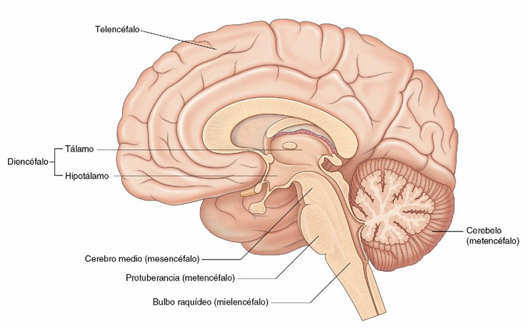
Fuente: Drake R, Vogl W, Mitchell A. Encéfalo corte sagital. GRAY. 2015
Los niños más pequeños tienen con más frecuencia tumores de origen embrionario, mientras que los de origen glial predominan en los más mayores. La patogénesis de la mayoría de los tumores cerebrales infantiles es desconocida. Se deben a síndromes genéticos hasta un 8% de los cánceres en la infancia y adolescencia. La exposición a radiaciones ionizantes es el único factor de riesgo ambiental asociado de manera consistente al desarrollo de tumores cerebrales en la infancia.
La clasificación de los tumores del SNC de la Organización Mundial de la Salud (OMS) de 2016, se basa en aspectos histológicos e incorpora además parámetros moleculares y genéticos. Estos avances han sido posibles gracias a estudios de genómica de alta resolución, epigenética y transcriptómica. Estos avances han posibilitado el desarrollo de terapias dirigidas a dianas moleculares específicas, especialmente relevantes en los gliomas, y la asignación más precisa de grupos de riesgo en el caso de los meduloblastomas y ependimomas.
MANIFESTACIONES CLÍNICAS:
La sintomatología es dependiente de la edad, de la localización del tumor, de si existe o no edema cerebral peritumoral acompañante y de la posible existencia de una obstrucción a la circulación del líquido cefalorraquídeo (LCR) que conduce a una hidrocefalia e hipertensión intracraneal (HTIC).
Los síntomas y signos más frecuentes son los derivados del aumento de la presión intracraneal (PIC), déficits neurológicos focales y epilepsia. Generalmente, la aparición de la sintomatología se acompaña de signos neurológicos anormales en la exploración clínica, aunque en los casos en que un tumor se presenta con crisis epilépticas (CE) es menos frecuente que la exploración sea anormal.
Es importante estar atentos a la progresividad de la sintomatología; la mayoría de los casos se diagnostican en los primeros 3-6 meses desde el inicio de los síntomas, aunque puede retrasarse especialmente en los casos que se presentan con crisis epilépticas o trastornos endocrinológicos. El tiempo hasta el diagnóstico se reduce en tumores de mayor agresividad y a menor edad del paciente.
- Signos y síntomas:
- Cefalea: constituye el síntoma más frecuente (50% de los niños la presentan antes del diagnóstico). Puede ser leve e intermitente, y responder a analgésicos. La cefalea persistente, que despierta o se presenta al despertar, es signo característico de HTIC. Es común que se acompañe de vómitos, repetidos, con mayor frecuencia por la mañana.
- Problemas de aprendizaje y del comportamiento (irritabilidad, letargia) son las formas de presentación en alrededor de un 10% de los casos cada una.
- Crisis epilépticas: son la forma de presentación en el 10-15% de los niños. Suelen ser crisis epilépticas focales motoras y focales con variaciones de conciencia. Son la manifestación más frecuente en algunos tipos de tumores, como astrocitomas de bajo grado y oligodendrogliomas de los hemisferios cerebrales.
- Signos anormales en la exploración: los más frecuentes son alteraciones de los pares craneales, papiledema y signos cerebelosos (cada uno de ellos en alrededor del 50% de los casos).
- Otras alteraciones características en la edad pediátrica son: macrocefalia, generalmente en lactantes, debida a HTIC; problemas de crecimiento (tanto de escasa ganancia de talla como de peso) y otros trastornos endocrinológicos, el más frecuente, la diabetes insípida.
- Otras formas habituales de presentación son las alteraciones visuales, además de diplopía por afectación del VI par, disminución de la agudeza visual asociada o no a déficits campimétricos (descritas estas dos últimas hasta en la mitad de los pacientes) y laterocolis o tortícolis asociado o no a rigidez de nuca en tumores de fosa posterior.
- En menores de 2 años, además de macrocefalia, retraso en la adquisición de hitos del desarrollo o pérdida de estos y alteraciones del comportamiento como irritabilidad. Otras formas de presentación: hemorragia intracraneal, náuseas, vómitos y retraso en el crecimiento, cuya forma más extrema es el síndrome diencefálico de emaciación.
- Otros signos no localizadores:
- Debidos a HTIC, aparte de la parálisis del VI par, afectación de los nervios craneales III (generalmente con midriasis unilateral exclusivamente), IV, V y VII, como consecuencia de la compresión de las fibras nerviosas contra estructuras óseas por la HTIC. Son más frecuentes en los tumores de fosa posterior.
- Ataxia, también puede aparecer en lesiones frontales.
- Signos y síntomas según la localización del tumor:
- Tumores supratentoriales: crisis epilépticas, signos neurológicos focales (el más frecuente, hemiplejia), alteraciones visuales, cognitivas y del comportamiento.
- Tumores cerebelosos: ataxia, movimientos oculares anormales, vértigo, estrabismo, rigidez de nuca, tortícolis o tortícolis, pérdida de peso, debilidad motora focal, síntomas auditivos.
- Tumores del tronco encefálico: ataxia, parálisis de pares craneales, signos piramidales, debilidad motora focal, parálisis facial, estrabismo, movimientos oculares anormales, problemas de aprendizaje y de conducta, vómitos intratables, retraso del crecimiento.
- Tumores diencefálicos y centrales (ependimarios): movimientos oculares anormales y estrabismo, disminución de la agudeza visual, diabetes insípida, ataxia, atrofia óptica, problemas de aprendizaje (problemas visuales, problemas de memoria, alteraciones de las funciones ejecutivas y de la coordinación motora fina), trastornos del comportamiento, alteración del nivel de conciencia, déficits campimétricos, crisis epilépticas, déficits motores focales, retraso del desarrollo psicomotor, talla baja, pérdida de peso, vértigo, síntomas auditivos.
- Tumores de la médula espinal: dolor de espalda (>50% de los casos), escoliosis y otras deformidades espinales (30% de los casos), trastorno de la marcha o de la coordinación (40%), debilidad motora focal, paraplejia, cuadriplejia, debilidad de miembros superiores, trastorno del esfínter (20%), laterocolis o tortícolis, alteraciones sensoriales, disfagia, cambios de la voz (estos últimos en lesiones de la unión cervicobulbar, a veces asociados a anomalías del patrón respiratorio, vómitos y cuadriplejia).
PRUEBAS COMPLEMENTARIAS:
Ante la sospecha de un tumor cerebral, la prueba de elección es el estudio de resonancia magnética (RMN) craneal pre y poscontraste junto al estudio de RMN espinal.
El rendimiento de la RMN se sitúa muy por encima de la tomografía axial computarizada (TAC), excepto para la visualización de calcificaciones intracraneales. La TAC sigue siendo la prueba de imagen más utilizada en situaciones de urgencia debido a que tiene la ventaja de no necesitar una anestesia general en pacientes no colaboradores, pero tiene a su vez los inconvenientes derivados de la exposición a radiación ionizante. Si no es posible disponer de RMN, la TAC debe hacerse pre y postinyección de contraste iodado para evitar falsos negativos.
El estudio del líquido cefalorraquídeo (LCR) no es generalmente esencial, salvo para aquellos tumores que pueden presentar diseminación leptomeníngea, en cuyo caso la citología es esencial para el estadiaje y ante la posibilidad de una enfermedad hematológica. El estudio de marcadores bioquímicos en LCR se utiliza en la valoración de los tumores de células germinales (gonadotropina coriónica humana y alfafetoproteína). La realización de una punción lumbar ante la sospecha clínica de una masa intracraneal debe realizarse siempre tras un estudio de neuroimagen y teniendo en cuenta las posibles contraindicaciones de esta técnica.
3.4.2 Meduloblastoma
El meduloblastoma (MB) es el tumor maligno más frecuente de la infancia. Es el segundo de los tumores de fosa posterior tras el astrocitoma cerebeloso. Su pico de incidencia es bifásico: 3-4 años y 8-9 años, y predomina en el sexo masculino. Suelen originarse en la región del vermis cerebeloso, en el techo del IV ventrículo y crecen hacia la luz del IV ventrículo y el dorso del vermis, produciendo hidrocefalia.
Las metástasis por diseminación a través del LCR son frecuentes. La presentación característica es con signos y síntomas de HTIC y ataxia de pocas semanas de evolución. Puede no haberse desarrollado papiledema en el momento de la presentación. La diseminación metastásica puede ocasionar dolor radicular y/o paraplejía. Puede dar lugar a múltiples neuropatías craneales.
En los estudios de RMN, el MB se caracteriza por ser un tumor sólido, de línea media, redondeado y con gran avidez homogénea por el gadolinio. Puede tener pequeñas áreas químicas, hemorragias o calcificaciones. La nueva clasificación de la OMS de 2016, basada en características moleculares e histológicas, reconoce 4 grupos moleculares:
- Wingless o WNT: Se asocia con el síndrome de Turcot. La supervivencia de este tipo de MB es superior al 95% a los 5 años.
- Sonic hedgehog (SHH): Se asocia al síndrome de Gorlin.
- Grupo 3.
- Grupo 4. Los MB de los grupos 3 y 4 presentan metástasis con frecuencia.
Estos grupos a su vez se dividen hasta constituir 12 subgrupos con un comportamiento biológico distinto.
Los tratamientos tradicionales para el meduloblastoma en niños mayores de 3 años incluyen una combinación de cirugía, radioterapia cráneo-espinal y esquemas de quimioterapia basados en platinos, independientemente del subtipo molecular.
La resección quirúrgica es el paso inicial en el tratamiento y se conoce que la resección total o casi total del tumor primario se correlaciona con mejores resultados, especialmente en pacientes con enfermedad no diseminada. Tras la resección máxima, los pacientes se estratifican convencionalmente en riesgo estándar o alto, en función de la extensión de la resección quirúrgica y el estado de diseminación en el momento del diagnóstico, dado por la RM cráneo-espinal del diagnóstico y el estudio del LCR. realizado como mínimo a los 15 días de la cirugía. La supervivencia libre de enfermedad a los cinco años es superior al 80% en aquellos de riesgo estándar, y del 60% al 65% en los de riesgo alto después de dicho tratamiento.
En los niños más pequeños, principalmente los que tienen menos de 3 o 4 años en el momento del diagnóstico, el tratamiento posquirúrgico suele realizarse con quimioterapia solamente.
3.4.3 Gliomas
Gliomas de alto grado:
El 20% de los gliomas infantiles son de alto grado. Representan de un 7 al 11% de los tumores cerebrales primarios en la edad pediátrica. Su incidencia es mayor en síndromes con predisposición al desarrollo de tumores como Li Fraumeni. Incluyen el astrocitoma anaplásico (AA) grado III de la OMS y el glioblastoma multiforme (GBM) grado IV de la OMS.
La forma más frecuente de presentación es con cefalea, hallazgos neurológicos focales y cambios de la personalidad. En un tercio de los casos se presentan con crisis epilépticas.
El pronóstico es desalentador, ya que más del 90% de los pacientes fallecen a la enfermedad en un plazo de 18 meses, con una supervivencia media de 9 meses. El tratamiento estándar incluye la radioterapia y ha sido el único tratamiento que ha alterado el curso de la enfermedad.
Gliomas de troncoencéfalo:
La presentación de los tumores de troncoencéfalo suele incluir una combinación de ataxia, neuropatías craneales y signos de vías largas. Los síntomas de HTIC generalmente son tardíos y puede no haber papiledema. Los vómitos son frecuentes. La irritabilidad y las alteraciones emocionales son comunes. Los tumores de lámina cuadrigémina o periacueductales producen hidrocefalia o síndrome de Parinaud.
El glioma difuso de la línea media se localiza de manera habitual en el puente. Son tumores grado IV de la OMS. En RMN son tumores grandes, de márgenes imprecisos, que expanden el puente y que característicamente se realzan poco o nada con gadolinio. Tienen muy mal pronóstico.
Los tumores situados en el bulbo y mesencéfalo son, con frecuencia, lesiones de bajo grado con componente exofítico (se proyecta hacia afuera de la superficie del tejido). Su pronóstico es mejor.
Gliomas de bajo grado:
Los gliomas de bajo grado son los tumores del SNC más comunes en los niños (40% de todos los tumores del SNC en menores de 18 años). Incluyen el:
- Astrocitoma pilocítico (AP), que constituye el tipo más frecuente.
- Astrocitoma pilomixoide.
- Astrocitoma de células gigantes subependimario (ACGSE).
- Xantoastrocitoma pilocítico.
- Astrocitoma difuso.
Todos ellos son grado I de la OMS, salvo el último que es grado II. La mayoría aparecen de forma esporádica, aunque el AP se asocia a la neurofibromatosis tipo I (NF1) y el ACGSE a la esclerosis tuberosa.
La clínica depende de la localización. La mayoría se encuentran en el cerebelo (fosa posterior). El síndrome diencefálico hipermetabólico de Rusell es característico de los tumores de localización hipotalámica, cursa con emaciación y retraso en el crecimiento a pesar de una ingesta calórica adecuada. Este síndrome también se puede originar por tumores de fosa posterior y craneofaringiomas.
El AP muestra un crecimiento lento y suele localizarse en los hemisferios cerebelosos. Son tumores bien definidos con componente quístico. Rara vez son infiltrantes. Los quistes pueden ser levemente hiperintensos con respecto al LCR. Se realzan intensamente con el gadolinio. El 10% pueden presentar diseminación leptomeníngea.
La resección quirúrgica es curativa para los gliomas de bajo grado, especialmente los AP, cuando son resecados totalmente. El reto terapéutico surge cuando los tumores aparecen cerca de estructuras vitales, lo que dificulta la resección sin causar una morbilidad o un compromiso funcional significativo. En tumores no resecables o con resección incompleta que requieren tratamiento se empleará la quimioterapia como primera opción, con la combinación de carboplatino y vincristina, o vinblastina semanal, intentando no emplear la radioterapia por los daños que pueden provocar a medio-largo plazo.
Gliomas de la vía óptica:
Representan el 4-8% de los tumores del SNC pediátricos. Aparecen especialmente en niños pequeños y son de lento crecimiento. La mayoría de los gliomas de la vía óptica son esporádicos, pero hasta un tercio se observan en pacientes que tienen una neurofibromatosis tipo I (NF1). Este tumor afecta al 20% de pacientes pediátricos con NF1. A pesar de ello no se recomienda el cribado sistemático en esta población si no hay síntomas, ya que exhiben un comportamiento menos agresivo a pesar de presentarse a edades más precoces. Tanto en el contexto de NF1 como en pacientes sin esta enfermedad se han descrito regresiones espontáneas.
Las lesiones anteriores al quiasma se presentan con proptosis y pérdida progresiva de visión monocular en unos meses. En la exploración podemos encontrar papiledema o atrofia óptica.
En niños con lesiones del quiasma o más posteriores se presentan con disminución de agudeza visual asimétrica o unilateral. En menores de 2 años se acompaña de nistagmo pendular adquirido, en ocasiones monocular. Los defectos de campos visuales habitualmente son asimétricos e irregulares.
El tumor puede extenderse al hipotálamo y dar lugar a hidrocefalia con o sin disfunción hipotálamo-hipofisaria.
3.4.4 Ependimomas
Los ependimomas son el tercer tumor por frecuencia y suponen el 15% de los tumores de fosa posterior. El 60% de los casos se presentan en menores de 5 años. En la mayoría de los niños se localizan en fosa posterior, sin embargo, en la adolescencia tienden a presentarse en los hemisferios cerebrales, no siendo rara la asociación a sangrado tumoral en estos casos.
Son tumores sólidos que se sitúan en línea media, ocupando el IV ventrículo, obstruyéndolo, y tienen tendencia a extenderse por los recesos laterales hacia la cisterna cerebelopontina y el canal espinal cervical. Pueden hacer metástasis por el LCR.
Las manifestaciones más frecuentes son síntomas/signos de HTIC y déficits focales. Las parálisis de nervios craneales, la rigidez de nuca y la tortícolis también son frecuentes.
El estándar de tratamiento sigue siendo la máxima resección quirúrgica segura, seguida de radioterapia postoperatoria adyuvante dirigida al sitio primario (en estos casos se administra a los mayores de un año porque es local).
3.4.5 Craneofaringioma
Constituye el 5-10% de los tumores cerebrales infantiles y el 50% de los tumores de línea media. Se pueden presentar a cualquier edad. Es un tumor epitelial benigno de lento crecimiento, originado en los remanentes celulares de la bolsa embrionaria de Rathke en el área supraselar adyacente al quiasma óptico. Por la localización suele presentarse con trastornos visuales uni o bilaterales como hemianopsia bitemporal, nistagmo, papiledema y atrofia óptica, y con problemas endocrinológicos.
Los pacientes habitualmente son diagnosticados meses después de su presentación por las anormalidades endocrinológicas como déficit de crecimiento, disfunción sexual, amenorrea, hipotiroidismo o diabetes insípida. Con frecuencia producen HTIC por hidrocefalia. No es infrecuente encontrar, además, somnolencia, apatía y cambios de humor.
Las lesiones suelen ser grandes en el momento del diagnóstico con calcificaciones en la región supraselar o infra y supraselar y la presencia de uno o varios quistes, que pueden comprimir el III ventrículo. Aunque se consideran tumores histológicamente benignos, siguen siendo un reto con respecto a las opciones de tratamiento.
3.4.6 Oligodendroglioma
Representan el 20% de los tumores supratentoriales. Son tumores de crecimiento lento. Tienen una importante tendencia a calcificar. La mayoría se localizan en el lóbulo frontal y se presentan con crisis epilépticas.
El pronóstico generalmente es bueno, incluso en pacientes con resección parcial, pues permanecen estables por largos períodos. Aunque, hay oligodendrogliomas malignos, de mal pronóstico.
3.4.7 Neuroblastoma
El neuroblastoma es el tumor sólido extracraneal más frecuente de la edad pediátrica. Es una enfermedad compleja y heterogénea donde diversos factores como: la edad al diagnóstico, el estadio, la localización del tumor primario y sus características histológicas y moleculares, determinan los pronósticos y condicionan el tratamiento.
El neuroblastoma es la neoplasia más frecuente en el primer año de vida, doblando en incidencia a la leucemia. Es el primer tumor sólido extracraneal diagnosticado entre 0-14 años de edad, ocupando el cuarto lugar en frecuencia de todas las neoplasias infantiles después de las leucemias, los tumores del sistema nervioso central y los linfomas.
La mediana de edad al diagnóstico es de 17-18 meses con una discreta predominancia en varones (ratio 1,2:1). Aproximadamente, el 30-40% de los casos se diagnostican durante el primer año de vida, el 90% antes de los 5 años de edad y el 98% antes de los 10 años. La enfermedad es muy rara en adolescentes y adultos jóvenes, en los que tiene un comportamiento más letal.
Los tumores neuroblásticos (neuroblastoma, ganglioneuroblastoma y ganglioneuroma) derivan de las células de la cresta neural (CN) comprometidas hacia el desarrollo del sistema nervioso simpático (SNS) y de las células gangliónicas de la médula adrenal. Su localización anatómica es diversa; se pueden originar a cualquier nivel en los ganglios simpáticos paravertebrales, desde el cuello hasta la pelvis, o en las glándulas suprarrenales.
Su presentación clínica es también variable, condicionada por la localización del tumor primario y la extensión de la enfermedad, presentando metástasis hasta el 50% de los pacientes al diagnóstico. Así, las familias pueden consultar con el pediatra de Atención Primaria o en el servicio de urgencias de Pediatría, por síntomas tan diversos como: una masa abdominal, estreñimiento, dolor abdominal, fiebre, dificultad respiratoria o síntomas neurológicos compatibles con compresión medular o, con menor frecuencia, un síndrome opsoclunus-mioclonus.
Su historia natural es también heterogénea. En neonatos y lactantes menores de 1 año de vida es, por lo general, un tumor de buen pronóstico. En cambio, en niños mayores de 18 meses, suele tener un pronóstico desfavorable a pesar del tratamiento multimodal intensivo.
Los avances en el estadiaje de la enfermedad, conseguidos gracias a las nuevas técnicas de imagen y de genética molecular, han facilitado la estratificación de los pacientes en grupos de riesgo con criterios definidos de tratamiento. Esto ha mejorado la supervivencia global a largo plazo, que se sitúa entre el 85-90%, para los pacientes de riesgo bajo o intermedio.
A pesar del tratamiento intensivo con quimioterapia, cirugía y radioterapia, seguidos de trasplante autólogo, ácido 13-cis retinoico e inmunoterapia, la supervivencia de los pacientes de alto riesgo no ha mejorado sustancialmente, siendo menor del 10% tras una caída o en pacientes con enfermedad refractaria. Y solo del 50% en las formas diseminadas de alto riesgo.
MANIFESTACIONES CLÍNICAS:
Pueden originarse en cualquier lugar del SNS, por lo que los signos y síntomas de presentación dependen de la localización del tumor primario, de su extensión y de la presencia o no de enfermedad diseminada. Mientras que los pacientes con neuroblastoma localizados suelen estar asintomáticos, los niños con enfermedad metastásica presentan síntomas sistémicos.
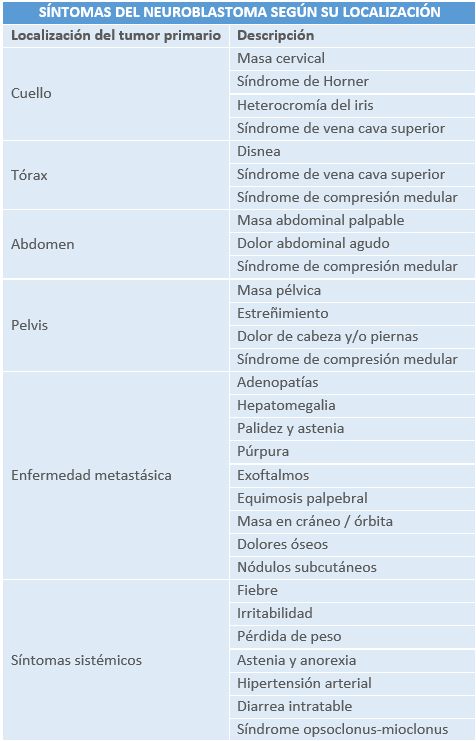
- Enfermedad localizada: el abdomen es el lugar de origen más frecuente (65% de los casos) con > 40% localizados en la glándula suprarrenal. En recién nacidos y lactantes es común el hallazgo casual de una masa suprarrenal en una ecografía. Los tumores abdominales de gran tamaño pueden causar: importante distensión abdominal, estreñimiento, dolor subagudo o agudo secundario a hemorragia intratumoral y/o hipertensión arterial (HTA) por compresión de los vasos renales. La producción de catecolaminas por el tumor también puede causar rubor, taquicardia e HTA.
- Otras localizaciones frecuentes son: cuello (5%), tórax (15%) y pelvis (5%). Los neuroblastomas cervicales pueden debutar con un síndrome de Horner por la lesión del ganglio estrellado o cervicotorácico. Los neuroblastomas torácicos generalmente son un hallazgo casual en una radiografía de tórax realizada en un paciente con tos persistente o dificultad respiratoria. Pueden comprimir y desplazar la vía aérea o el parénquima pulmonar y, ocasionalmente, producir un síndrome de vena cava superior. A nivel pélvico, pueden causar síntomas compresivos, como retención urinaria y estreñimiento. Cuando se originan en los ganglios paraespinales, pueden presentar síntomas neurológicos compatibles con un síndrome de compresión medular (dolor de espalda, debilidad muscular, alteraciones sensitivas), al extenderse a lo largo de las raíces nerviosas y a través del agujero neural.
- Enfermedad metastásica: alrededor del 55% de los pacientes presentan metástasis al diagnóstico (80% de los niños mayores y 40% de los lactantes). La enfermedad se disemina a los ganglios linfáticos locorregionales o por vía hematógena a huesos y médula ósea. Las metástasis óseas causan: dolor, irritabilidad, inflamación y cojera. La infiltración medular puede causar anemia y trombocitopenia. Las metástasis en hígado y piel son más frecuentes en lactantes menores de 18 meses de edad.
- Enfermedad 4S o MS: Es una forma de presentación especial en lactantes con metástasis limitadas a hígado, médula ósea y piel, y un tumor primario de pequeño tamaño y con tendencia a la regresión espontánea. La infiltración hepática masiva puede progresar rápidamente y causar insuficiencia hepática y coagulopatía junto con una gran distensión abdominal. Esta puede producir dificultad respiratoria, insuficiencia renal y edema escrotal y de miembros inferiores, por compresión de la vena cava inferior, especialmente en menores de 3 meses de edad.
- Síndromes paraneoplásicos: generalmente, asociados a neuroblastomas localizados y diferenciados. La secreción de péptido intestinal vasoactivo por el tumor puede causar una diarrea secretora intratable. El síndrome opsoclonus-mioclonus se presenta en el 2-3% de los pacientes y, aunque los pronósticos del tumor suelen ser favorables, el 70-80% de los niños presentarán déficits neurológicos a largo plazo.
DIAGNÓSTICO:
El diagnóstico del neuroblastoma se basa en la combinación de varias pruebas: estudios de imagen, marcadores tumorales, catecolaminas en orina y el análisis histológico y genético del tumor.
La biopsia del tumor primario o de una lesión metastásica de partes blandas es necesaria para el diagnóstico. En pacientes inestables en los que el riesgo quirúrgico no es asumible, se puede realizar el diagnóstico demostrando la infiltración tumoral en médula ósea y la elevación de catecolaminas en orina. Las catecolaminas se encuentran elevadas en la orina en el 90% de los niños con neuroblastoma.

Además de las radiografías convencionales y ecografías iniciales, se debe evaluar el tumor primario mediante RNM o TAC, determinando su localización, extensión, afectación ganglionar, relación con estructuras vecinas y diseminación metastásica. Se incluye el tumor primario, así como tórax, abdomen y pelvis.
También se realizarán aspirados y biopsias de médula ósea bilaterales al diagnóstico y a intervalos regulares durante el tratamiento y en el seguimiento de los pacientes de alto riesgo.
TRATAMIENTO:
La cirugía tiene un papel fundamental en el neuroblastoma, tanto en el diagnóstico como en el tratamiento.
El abordaje quirúrgico, ya sea mediante cirugía abierta o técnicas menos invasivas (laparoscopia, toracoscopia, biopsia percutánea), dependerá de la localización, extensión y resecabilidad de la lesión. Los tumores localizados sin factores de riesgo definidos por imagen (IDRF) en el neuroblastoma se resecarán en la cirugía inicial. En los demás casos, la biopsia garantizará la obtención del material suficiente para realizar todos los estudios y se valorará extirpar el resto tumoral de forma diferida. El riesgo de complicaciones quirúrgicas es mayor en los lactantes, sobre todo en menores de 2 meses de edad, en los que la enfermedad tiene mejores pronósticos; por lo que, en estos casos, se debe valorar cuidadosamente la indicación de la cirugía.
El neuroblastoma es un tumor radiosensible, pero la elevada prevalencia de metástasis óseas hace que la radioterapia no sea curativa. La radioterapia está indicada como tratamiento de la enfermedad residual en algunos tumores localizados, en el control de la enfermedad local y de metástasis refractarias en pacientes de alto riesgo, así como en pacientes en fase terminal, para mitigar el dolor asociado a las metástasis óseas. Actualmente, el uso de radioterapia urgente en la compresión medular o en la infiltración hepática masiva es excepcional, siendo preferible la quimioterapia, tanto por su eficacia como por el menor riesgo de efectos secundarios a largo plazo.
La quimioterapia está indicada en pacientes de riesgo intermedio o alto, así como en algunos pacientes de bajo riesgo con compromiso vital, variando la intensidad de la misma según el riesgo.
El tratamiento del neuroblastoma depende del grupo de riesgo al que pertenezca el paciente, basado en las diferencias pronósticas relacionadas con la edad y la biología tumoral, por lo que las estrategias terapéuticas son muy variadas.
- Tratamiento del neuroblastoma de bajo y muy bajo riesgo:
Su pronóstico es excelente, con Supervivencia Libre de Enfermedad (SLE) > 90% y supervivencia global (SG) cercana al 100%. La mayoría de estos pacientes se curan solo con cirugía y en algunos casos sin ningún tratamiento, dada la tendencia de estos tumores a la regresión espontánea.
La quimioterapia se reserva para los pacientes con síntomas amenazantes para la vida, como compresión medular o distrés respiratorio secundario a infiltración hepática masiva.
Los fármacos utilizados son: etopósido, carboplatino, ciclofosfamida y doxorrubicina, y se administrarán sólo los ciclos necesarios para el control de los síntomas.
- Tratamiento del neuroblastoma de riesgo intermedio:
Es un grupo muy heterogéneo con tumores de características biológicas variables, pero con tasas de SG > 80%, cuando se combinan quimioterapia y cirugía.
- Tratamiento del neuroblastoma de alto riesgo:
Los pacientes de alto riesgo reciben tratamiento multimodal intensivo con: quimioterapia, cirugía, radioterapia, inmunoterapia y terapia diferenciadora con ácido 13-cis retinoico. Presentan caídas frecuentes y alta mortalidad, con una probabilidad de SG a los 5 años del 50%.
El tratamiento se divide en 3 fases:
- Inducción (quimioterapia y cirugía)
- Consolidación (trasplante autólogo y radioterapia)
- Mantenimiento (inmunoterapia y ácido 13-cis-retinoico)
El objetivo de la quimioterapia de inducción es conseguir la máxima reducción del tumor primario y de las metástasis. La intensidad de la dosis se relaciona con la respuesta y la supervivencia. El régimen recomendado es: vincristina, carboplatino, etopósido, cisplatino y ciclofosfamida. Se administra en un periodo de tiempo muy ajustado (3-4 meses) y trata de evitar el desarrollo de resistencias farmacológicas.
En los pacientes con respuesta adecuada, se procede a la extirpación quirúrgica del tumor primario.
Los demás pacientes continúan con la fase de consolidación, que consiste en quimioterapia mieloablativa (busulfán y melfalán) seguida de rescate con trasplante autólogo y radioterapia local. El tratamiento de mantenimiento incluye ácido 13-cis-retinoico, que induce la diferenciación de las células tumorales residuales e inmunoterapia con el anticuerpo monoclonal anti-GD2, el dinutuximab.
3.4.8 Tumores óseos
Osteosarcoma o sarcoma osteogénico
El osteosarcoma es un tumor maligno primario del hueso, de origen mesenquimal, compuesto por células neoplásicas productoras de osteoide.
Representa el 55% de los tumores óseos en menores de 20 años y en adolescentes es más frecuente que el Sarcoma de Ewing, con una incidencia máxima entre 13 y 16 años de edad. Por razones desconocidas, el osteosarcoma afecta más a los varones (proporción 1,34 a 1) y es más frecuente en la raza negra y en otras razas en comparación con los caucásicos. Se localiza preferentemente en las metáfisis de los huesos largos, especialmente: fémur distal, tibia proximal y húmero proximal.
Muchas veces su aparición se relaciona con un traumatismo reciente, pero no es cierto que este sea la causa, sólo pone de manifiesto la enfermedad. La exposición a radiación ionizante sí que es un agente causal verificado, con períodos de latencia de 10-20 años. También se relaciona con la aparición de osteosarcoma la administración previa de citostáticos alquilantes. Se han descrito asociaciones con enfermedades hereditarias, la más clara con el retinoblastoma hereditario; también con los síndromes de Li-Fraumeni, Rothmund-Thomson, Bloom y la enfermedad de Paget.
CLASIFICACIÓN:
El tipo histológico más frecuente es el ostosarcoma convencional (intramedular de alto grado, 90% de todos los ostosarcomas), con tres subcategorías en función del componente celular predominante: osteoblástico (50%), fibroblástico (25%) y condroblástico (25%). Además de estas 3 subcategorías, se describen dos variantes: ostosarcoma telangiectásico (quístico, vascularizado) y ostosarcoma de célula pequeña, muy agresivo y morfológicamente similar al sarcoma de Ewing.
Otra categoría son los ostosarcomas superficiales, en contraposición a los intramedulares, que incluyen: tipo parostal de bajo grado, tipo perióstico de grado intermedio y osteosarcomas superficiales de alto grado. En general, son menos agresivos y su evolución más favorable.
MANIFESTACIONES CLÍNICAS:
El principal síntoma es el dolor, localizado en una extremidad con más frecuencia y, generalmente, lleva varios meses de evolución antes del diagnóstico.
El paciente lo suele relacionar con el ejercicio físico o un traumatismo. Con el tiempo, aparecerán daños locales, efectos de masa e impotencia funcional. No suele haber fiebre, pérdida de peso ni otra sintomatología sistémica.
El osteosarcoma tiene predilección por las metáfisis de los huesos largos: el 80% se localiza en las extremidades (fémur 40%, tibia 20%, húmero 10%), creciendo desde la cavidad medular hacia la corteza y los tejidos blandos. Las metástasis aparecen, sobre todo, en el pulmón, sin clínica acompañante.
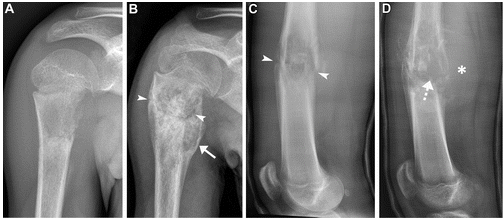
Fuente: Nguyen J, et al. Cambios radiográficos en respuesta a la quimioterapia neoadyuvante en dos pacientes. RadioGraphics. 2022.
DIAGNÓSTICO:
El diagnóstico del tumor es anatomopatológico y es necesario realizar siempre un estudio de extensión buscando metástasis.
El osteosarcoma aparece en la radiografía como una masa heterogénea, con zonas osteolíticas y escleróticas, de bordes mal definidos, que crece desde la cavidad medular y progresa hacia la corteza, atravesando y levantando el periostio (que reacciona formando tejido óseo inmaduro, en forma de triángulo: signo de Codman), y pudiendo afectar a los tejidos blandos que rodean al hueso, produciendo imágenes difusas de diferentes densidades. Para valorar la extensión del tumor (ósea y en partes blandas) y la afectación de estructuras contiguas (vasos, nervios…) es necesario realizar una resonancia magnética (RNM) o una tomografía computarizada (TAC), siendo preferible la RM por su mayor sensibilidad. En ocasiones, existen lesiones satélites (skip) en el propio hueso, sin contigüidad con el tumor principal, por lo que hay que incluir la totalidad del afecto óseo en la técnica de imagen.
El estudio histopatológico (biopsia) del tumor proporciona el diagnóstico de certeza. Debe obtenerse mediante un trócar y dentro de la zona que se marcará, para después ser resecada con el tumor.
Conocido el diagnóstico anatomopatológico, se realiza el estudio de extensión, es decir, se completará el estadiaje del tumor con un TAC pulmonar de alta resolución, para detectar metástasis pulmonares, y una gammagrafía ósea con tecnecio (99Tc) para localizar metástasis óseas. La tomografía por emisión de positrones (PET) se está usando cada vez más para valorar la afectación metastásica, principalmente si se combina con TAC.
Aunque existen varias clasificaciones para el estadiaje, realmente es suficiente con distinguir entre tumor localizado (L) o con metástasis (M). En el momento del diagnóstico, el osteosarcoma se presenta como enfermedad metastásica en el 20% de los casos.
TRATAMIENTO:
El abordaje debe ser multidisciplinar, con la administración de quimioterapia antes y después de la cirugía, siendo esencial conseguir la resección en bloque de todo el compartimento tumoral.
Debido a la alta incidencia de micrometástasis, indetectables, pero presentes ya en el momento del diagnóstico, la cirugía por sí sola no consigue curar más allá del 20% de los pacientes. Por ello, resulta esencial administrar quimioterapia adyuvante para erradicarlas.
- Quimioterapia neoadyuvante en ciclos, combinando metotrexato a altas dosis, cisplatino y doxorrubicina. Además de eliminar las micrometástasis, consigue una disminución de volumen del tumor facilitando así su resección.
- Cirugía, que debe conseguir una resección completa del tumor en bloque, incluyendo: el compartimento óseo, las partes blandas circundantes y los trayectos cutáneos y subcutáneos de las biopsias iniciales, y con márgenes de seguridad. La reconstrucción del sistema musculoesquelético se consigue mediante injertos óseos (autólogos o alogénicos) o prótesis. Son cirugías complejas que requieren ser realizadas por un cirujano ortopédico con experiencia en el tratamiento de tumores óseos pediátricos. Actualmente, se consigue salvar la extremidad afecta en el 80% de los casos.
- Quimioterapia postquirúrgica con los mismos agentes o agregando ifosfamida, para eliminar las células tumorales residuales que hayan podido movilizarse con la intervención.
Con esta estrategia se ha conseguido elevar la supervivencia libre de eventros (SLE) a los 5 años hasta el 60-70%, pero desde 1990, no se han conseguido mejorías significativas.
El osteosarcoma no es un tumor muy radiosensible, por lo que la radioterapia solo se emplea en localizaciones axiales en las que la cirugía no es posible, en casos de osteosarcoma multifocal o con fines paliativos para el control del dolor.
Para la enfermedad metastásica se emplean combinaciones de quimioterapia con los mismos citostáticos, pero con mayor número de ciclos, además de resecar el tumor primario y las metástasis pulmonares que continúen siendo visibles tras la quimioterapia neoadyuvante. A pesar de todo ello, el SLE oscila entre un 16-53%.
Tumores de la familia Ewing
El Sarcoma de Ewing es el segundo tumor óseo maligno primario por frecuencia en niños y adolescentes, siendo el tumor óseo más frecuente en niños menores de 4 años. Es raro por encima de los 30 años, y en las razas negra y asiática. Predomina en varones (1,5:1). No es hereditario ni se asocia a síndromes malformativos, y tampoco se relaciona con ningún agente externo causal.
Los Tumores de la Familia Ewing comparten la misma anomalía cromosómica, consistente en una translocación entre los cromosomas 11 (gen EWS) y 22 (gen FLI1), originando el gen quimérico EWS-FLI1.
La proteína resultante ocasiona una desregulación en los genes responsables de la proliferación y diferenciación celular, favoreciendo el desarrollo del tumor. Se detecta en el 90-95% de los Tumores de la Familia Ewing y, por ello, es un marcador de diagnóstico que ayuda a distinguir el Sarcoma de Ewing de otras entidades.
Todos los Tumores de la Familia Ewing expresan niveles altos de una glicoproteína de membrana (en concreto: CD99) que ayuda a distinguirlos de otros tumores de células redondas y pequeñas de la infancia (neuroblastoma, otros sarcomas, linfomas).
CLASIFICACIÓN:
Se compone de:
- Sarcoma de Ewing típico y atípico
- Tumor neuroectodérmico primitivo (PNET, anteriormente llamado neuroepitelioma)
- Tumor de células pequeñas de la región toraco-pulmonar (tumor de Askin)
- Sarcoma de Ewing extraóseo
MANIFESTACIONES CLÍNICAS:
Los Tumores de la Familia Ewing pueden originarse en cualquier hueso y en tejidos blandos. Son algo más frecuente en el esqueleto axial (54%), sobre todo en los huesos de la pelvis (25%). En las extremidades, el lugar más común es el fémur (16%). El dolor local suele ser el síntoma inicial (96%); al comienzo tiene carácter intermitente y paulatinamente gana intensidad. Suele haber afectación de los tejidos blandos adyacentes, apareciendo tumefacción local. A veces, existen síntomas generales, como fiebre (21%). Otra sintomatología dependerá de la localización del tumor: derrame pleural en los tumores torácicos, dolor radicular en tumores vertebrales y problemas de esfínteres en tumores pélvicos.
DIAGNÓSTICO:
En la radiografía simple, el hueso afecto presenta un patrón moteado difuso, con predominio de áreas líticas. Puede existir el triángulo de Codman, y es típica la imagen en “capas de cebolla”, debido a la existencia de múltiples capas de reacción perióstica con neoformación ósea. En los huesos planos predominan zonas de esclerosis. La RNM es la técnica radiológica de elección para valorar la extensión ósea y extraósea del tumor. La TAC proporciona información sobre la cortical y los cambios en la estructura ósea.
El estudio anatomopatológico proporciona el diagnóstico mediante biopsia, similar al definido en el osteosarcoma.
Los Tumores de la Familia Ewing tienen una elevada capacidad de metastatizar a distancia en: pulmón (38%), huesos (31%) y médula ósea (11%). En el momento del diagnóstico, un 20% de los pacientes presentan metástasis visibles, pero la mayoría tienen metástasis subclínicas. Como en el osteosarcoma, es preciso realizar una TAC torácica de alta resolución y una gammagrafía ósea (99Tc). Además, se descartarán las metástasis en médula ósea, obteniendo, al menos, dos biopsias en dos lugares alejados del tumor primario. Como para el osteosarcoma, para el estadiaje es suficiente con distinguir entre tumor localizado (L) o con metástasis (M).
TRATAMIENTO:
La quimioterapia sistémica es imprescindible para curar un Tumores de la Familia Ewing, dada la presencia de micrometástasis desde el comienzo. En los últimos años, la combinación de ciclofosfamida, vincristina y doxorrubicina, alternando con ifosfamida y etopósido, se ha impuesto como la más eficaz y con menos toxicidad que otras combinaciones.
El esquema de tratamiento es similar al del osteosarcoma:
- Quimioterapia inicial neoadyuvante, para reducir el volumen tumoral y eliminar las micrometástasis.
- El tratamiento local consistirá en la resección quirúrgica, siempre que sea posible, con los mismos criterios que para el osteosarcoma. Los Tumores de la Familia Ewing son radiosensibles y, si el hueso no es resecable, o la cirugía supone una grave mutilación o deformidad estética, se utiliza la radioterapia como tratamiento local único.
- En la fase de consolidación tras el tratamiento local, se administra más quimioterapia para eliminar tumor residual y radioterapia posquirúrgica, si el grado de necrosis tumoral no es superior al 90%, o existen células tumorales en los márgenes de la resección.
En tumores de mal pronóstico tras la cirugía, se puede optar por una quimioterapia mieloablativa con rescate de progenitores hematopoyéticos autólogos. Para el acondicionamiento, se utiliza la combinación de busulfán y melfalán. En caso de persistir metástasis pulmonar, al final del tratamiento se puede administrar radioterapia pulmonar.
Rabdomiosarcoma
El rabdomiosarcoma es un tumor mesenquimal maligno que se asemeja al músculo estriado. Representa aproximadamente la mitad de los diagnósticos de sarcoma de partes blandas en Pediatría. La incidencia anual es entre 4 y 5 casos por millón.
Aunque se han descrito factores de riesgo, tanto ambientales, como genéticos, el rabdomiosarcoma es, como la mayoría de los tumores pediátricos, una neoplasia sin posibilidad de prevención.
CLASIFICACIÓN:
Desde el punto de vista anatomopatológico, se clasifica en cuatro variantes morfológicas, de las que tres aparecen en la edad pediátrica:
- Embrionario (80%)
- Alveolar (20%)
- Fusocelular/esclerosante (menos del 1%).
El rabdomiosarcoma embrionario se caracteriza por su aspecto inmaduro, mientras que el alveolar se distingue por la observación de septos rodeando formaciones alveolares tumorales.
MANIFESTACIONES CLÍNICAS:
La presentación clínica es variable, dependiendo de la ubicación del tumor. Puede manifestarse por el efecto funcional de la masa (tumefacción, dolor, impotencia funcional) o, en ocasiones, detectarse en una exploración clínica o radiológica rutinaria.
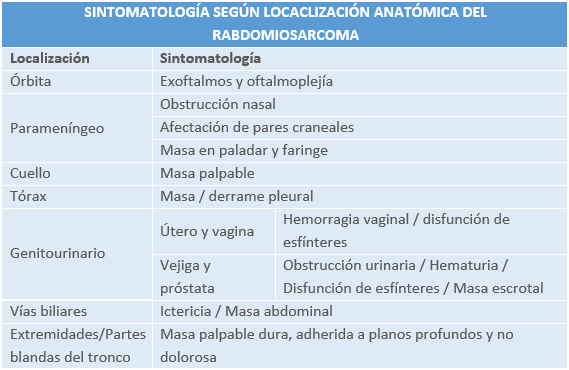
DIAGNÓSTICO:
La evaluación de la imagen radiografía simple inicial se completará con imagen de alta resolución mediante RNM del tumor primario, para valorar la extensión del tumor y su relación con las estructuras vecinas.
Es fundamental el estudio anatomopatológico del tumor mediante biopsia para establecer el subtipo histológico y, además, hay que determinar la presencia de casos que se clasifican como de alto riesgo y su tratamiento será más agresivo.
Para el estudio de extensión, la búsqueda de metástasis incluye un TAC torácica de alta resolución y una gammagrafía ósea (con 99Tc). En pacientes de alto riesgo (alveolares, FOXO positivo, metastásticos), se recomienda completar el estudio con biopsia bilateral de médula ósea. En los últimos años, la 18F-FdG-PET-TC con contraste está reemplazando al estudio de extensión tradicional, especialmente al diagnóstico, sin haber podido aún establecer su papel en la evaluación de respuesta.
TRATAMIENTO:
Todos los pacientes necesitan la administración de quimioterapia sistémica, incluso los resecados completamente, ya que se considera una enfermedad diseminada: el estudio por biopsia líquida detecta ADN tumoral en sangre periférica en 2/3 de los casos localizados. La quimioterapia se basa en la combinación de vincristina, actinomicina y ciclofosfamida/ifosfamida, con la introducción en los últimos años de irinotecán. El uso de antraciclinas no parece aumentar la supervivencia en pacientes de riesgo estándar/alto y se reserva solo para casos de riesgo muy alto (metastásicos y FOXO +).
El control local es clave en el rabdomiosarcoma. Dado que puede localizarse en muy diferentes ubicaciones, la cirugía requiere la participación de diferentes especialistas, además de cirujanos pediátricos: oftalmólogos, cirujanos maxilofaciales, otorrinolaringólogos, urólogos… El control de la enfermedad local idealmente debe obtenerse mediante la cirugía, pero la radioterapia es eficaz y puede considerarse como terapia local única en casos inoperables o con un riesgo quirúrgico inaceptable. El rabdomiosarcoma es una de las indicaciones aprobadas de protonterapia. La radioterapia local se administra concomitante con la quimioterapia de consolidación, mientras que la irradiación de las metástasis pulmonares se demora tras esta.
El tratamiento en Europa se basa en los protocolos elaborados por el Grupo Europeo Pediátrico de Sarcomas de Tejidos Blandos (EpSSG: European pediátrico Softissue Sarcoma Group). Se estratifica a los pacientes en grupos de riesgo y se establece el tratamiento:
- Los pacientes de riesgo bajo, con resección completa inicial y genética favorable, dado que ya se ha obtenido el control local, reciben únicamente quimioterapia de consolidación.
- Los pacientes de riesgo intermedio (representan el grupo más numeroso) se tratan, en su mayoría, de casos con biopsia inicial o resección incompleta, y genéticamente favorable. Reciben quimioterapia de inducción, seguida de control local (cirugía ± radioterapia) y de nueva quimioterapia de consolidación.
- Los pacientes de riesgo alto/muy alto presentan reordenamiento de FOXO y/o enfermedad metastásica. El esquema terapéutico es similar al riesgo intermedio, con las siguientes consideraciones: se recomienda la combinación de cirugía y radioterapia para el control local, y se añade, tras la consolidación, una fase de quimioterapia de mantenimiento (combinando ciclofosfamida oral y vinorelbina intravenosa) durante un mínimo de 6 meses.
Tumor de Wilms
El tumor renal más frecuente (90%) es el tumor de Wilms, con una prevalencia de 1 caso entre 10.000 niños menores de 15 años. Otros tumores renales menos frecuentes son: nefroma mesoblástico, sarcoma de células claras, tumor rabdoide-teratoide atípico y carcinoma renal.
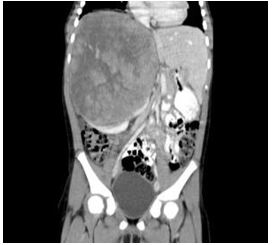
Fuente: Alonso Quintela P, et al. Resonancia magnética tras inyección de gadolinio, corte coronal. Anales de Pediatría. 2011.
El tumor de Wilms aparece con una edad típica de presentación entre los 3 y 5 años de edad. Se desarrolla a partir de restos nefrogénicos o tejido metanéfrico persistente. Estos restos están presentes en el 1% de los niños al nacimiento, pero suelen regresar durante la infancia. Se reconocen en todos los casos de tumor de Wilms bilateral y en el 35% de los tumores unilaterales y se consideran lesiones precursoras.
Los pronósticos del tumor de Wilms han mejorado en los últimos años, variando según el estadio y la histología, con unos porcentajes de supervivencia a 5 años del 85-90% en los casos favorables. Los pacientes con estadios avanzados y con histología desfavorable tienen peores pronósticos, con una supervivencia a 5 años del 38-84%)
CLASIFICACIÓN:
Los tumores de Wilms se dividen en 3 grupos de riesgo en función de la histología:

Tras la cirugía, se estratifica el riesgo según el subtipo histológico y el estadio local. Estos datos, en combinación con el volumen tumoral inicial, permiten clasificar a los pacientes en grupos de riesgo.

MANIFESTACIONES CLÍNICAS:
La presentación clínica más frecuente del tumor de Wilms es el hallazgo casual de una masa abdominal a la exploración sin otros síntomas acompañantes. Puede ser detectada en la exploración rutinaria por el pediatra o por los propios padres al vestir o bañar al niño.
El dolor se da en un 30-40% de los casos y la hematuria, que es menos frecuente, se presenta de forma intermitente como microhematuria (24%) o, en ocasiones, macrohematuria (18%). También puede presentarse de forma más infrecuente con afectación del estado general: fiebre, vómitos y otros síntomas constitucionales.
La tríada clásica de dolor, hipertensión y hematuria es poco frecuente en este tipo de tumores, apareciendo en aproximadamente una cuarta parte de los pacientes. La hipertensión se debe a la secreción de renina por parte del tumor, aunque también puede darse por compresión de vasos renales.
A la palpación, se trata de una masa generalmente de bordes bien definidos que se localiza en uno de los flancos. Rara vez, sobrepasa la línea media abdominal. Una vez detectada, hay que realizar la exploración física de manera cuidadosa, puesto que se corre el riesgo de presentar una rotura tumoral.
Una forma de presentación más infrecuente es en forma de abdomen agudo, tras la rotura del tumor. Esto puede suceder de forma espontánea o tras un traumatismo abdominal, y cursa con hemoperitoneo, siendo una urgencia quirúrgica.
En el diagnóstico de tumor de Wilms, hay que buscar los signos clínicos típicos de síndromes de predisposición genética, por si no hubieran sido detectados previamente.
La diseminación tumoral se puede producir por contigüidad a través de la cápsula renal, o de una forma hematógena. Aunque presentar metástasis al diagnóstico es poco frecuente (12% de los casos), en caso de aparecer, la localización más frecuente es el pulmón (80%). Otras localizaciones metastásicas menos frecuentes son: el hígado (15%), y aún más raras, el hueso, la médula ósea y el sistema nervioso central.
DIAGNÓSTICO:
Inicialmente, y dada la rápida disponibilidad de la misma, la primera prueba diagnóstica a realizar tras la exploración es una ecografía.
Mediante la ecografía, se puede determinar si la masa se origina en el riñón. También permite determinar la presencia de trombos intravasculares o de infiltración vascular por parte del tumor mediante el empleo de Doppler. La limitación principal de esta prueba es que es operador dependiente, y que puede verse alterada por la presencia de gas en el abdomen o por obesidad.
Una vez detectada una masa renal, para determinar la anatomía de la misma y su extensión, se debe realizar otra prueba de imagen, para lo cual la mayor parte de niños necesita sedación. Preferiblemente, se realizará una RMN abdominal. En caso de que esta prueba no esté disponible en un tiempo razonable o si se trata de un abdomen agudo, se puede realizar también una TAC abdominal (siendo preferible la RMN para evitar la radiación). Con estas pruebas, además de la extensión y naturaleza de la masa detectada, se puede detectar la presencia de otras masas abdominales y determinar si existe afectación del riñón contralateral.
Además, se debe realizar una radiografía basal de tórax, así como una TAC torácica para detectar metástasis pulmonar.
En los casos en los que la clínica, la edad del paciente y la imagen radiológica son claramente sugestivas de tumor de Wilms, en Europa, generalmente se inicia tratamiento con quimioterapia preoperatoria sin confirmación histológica.
Sin embargo, en los casos en los que existan dudas tras realizar las pruebas diagnósticas pertinentes, puede ser necesaria la realización de una extirpación quirúrgica total de la masa para determinar la naturaleza de la misma, en caso de ser posible. En el caso de tratarse de una masa irresecable, puede ser necesaria la realización de una biopsia.
Los pacientes con otro tipo de tumores renales pueden necesitar otro tipo de pruebas como son la RMN cerebral y la gammagrafía ósea en el tumor rabdoide y en el sarcoma de células claras.
TRATAMIENTO:
Actualmente, en Europa, se siguen las directrices de tratamiento del protocolo SIOP-UMBRELLA-RTSG2016 (Renal Tumor Study Group of the International Society of Pediatric Oncology), mientras que, en los países americanos, se tratan según el protocolo COG (Children’s Oncology Group). La principal diferencia entre ambos es la cirugía inicial que recomienda COG, mientras que en SIOP, se administra quimioterapia precirugía con el objetivo de reducir las posibles complicaciones derivadas de la misma, principalmente rotura tumoral, con la consiguiente menor necesidad de radioterapia. Ambos protocolos son comparables en términos de supervivencia.
De acuerdo con el protocolo SIOP, se administra quimioterapia preoperatoria a todos los pacientes entre los 6 meses y 16 años de edad. En niños menores de 6 meses es frecuente el nefroma mesoblástico congénito, cuyo tratamiento es únicamente quirúrgico. Por ello, en esta edad, se debe valorar individualmente el riesgo de rotura tumoral frente a la toxicidad de la quimioterapia. En pacientes mayores de 16 años, se debe descartar carcinoma de células renales mediante cirugía inicial, siempre que sea posible.
Para tumores localizados, la quimioterapia preoperatoria consiste en la combinación de vincristina y actinomicina durante 4 semanas. La cirugía se debe realizar entre las semanas 5 y 6, y está recomendada la nefrectomía radical con toma de muestras de adenopatías. En los metastásicos se añade doxorrubicina y el tratamiento inicial se prolonga 6 semanas. Con este régimen, el 61-67% de los pacientes presentan remisión de las metástasis previamente a la cirugía.
El tratamiento posquirúrgico, según estadio y subtipo histológico, es el siguiente:
- Estadio I:
- Histología de bajo riesgo: no requiere tratamiento posterior.
- Histología de riesgo intermedio: vincristina y actinomicina 4 semanas.
- Histología de alto riesgo: vincristina, actinomicina y doxorrubicina 27 semanas.
- Estadios II y III:
- Histología de riesgo bajo/intermedio: vincristina y actinomicina 27 semanas.
- Histología de riesgo alto: régimen HR1 34 semanas (combina ciclofosfamida, doxorrubicina, etopósido y carboplatino).
- En caso de enfermedad metastásica al diagnóstico, el tratamiento posquirúrgico combina vincristina, actinomicina y doxorrubicina, además de radioterapia pulmonar en caso de nódulos mayores de 3 mm. Siempre que sea posible, se recomienda la extirpación de nódulos para la confirmación histológica. Los nódulos menores de 3 mm no se consideran metástasis.
- En el caso de estadio IV en pacientes con histología de alto riesgo, se recomienda un régimen basado en: vincristina, irinotecán, ciclofosfamida, carboplatino, etopósido y doxorrubicina, seguido de “megadosis” de quimioterapia con rescate autólogo de progenitores hematopoyéticos.
En la enfermedad bilateral, uno de los objetivos primordiales es preservar la mayor funcionalidad renal, evitando en lo posible la nefrectomía total. El tratamiento inicial es vincristina y actinomicina durante 6 semanas con reevaluaciones periódicas hasta poder realizar cirugía. Si no se observa respuesta después de 12 semanas de tratamiento, se cambia el régimen quimioterápico a carboplatino/etopósido. La cirugía debe intentar preservar el mayor tejido funcional posible (“cirugía conservadora de nefronas”). El tratamiento posquirúrgico debe ser el correspondiente a la histología de más riesgo y el estadio más avanzado.
Cáncer de tiroides
Aproximadamente el 2% de los niños tienen nódulos tiroideos palpables. La mayoría son benignos, incluyendo lesiones inflamatorias o adenomas foliculares, pero hasta el 25% son malignos.
La glándula tiroides en los niños es particularmente susceptible a la irradiación y la carcinogénesis, lo que puede explicar por qué los niños con cáncer de tiroides tienden a presentar enfermedad avanzada. En comparación con los adultos, los niños con cáncer de tiroides muestran una mayor frecuencia de metástasis en los ganglios linfáticos y metástasis a distancia en el momento del diagnóstico y, además, tasas más altas de recidiva. A pesar de estas características, generalmente tienen un buen pronóstico.
CLASIFICACIÓN:
La mayoría de los nódulos tiroideos en los niños son benignos. Las causas incluyen adenomas tiroideos benignos (por lo general, adenomas coloidales o foliculares), quistes tiroideos y lesiones inflamatorias. Los adenomas foliculares son el tumor más común.
Los tipos de tumores malignos de tiroides son:
- Cánceres de tiroides diferenciados (CDT):
- Cáncer papilar de tiroides (PTC): El carcinoma papilar representa aproximadamente el 86% de los cánceres de tiroides pediátricos, con fuertes aumentos en este tipo durante las últimas dos décadas. Una "neoplasia folicular tiroidea no invasiva con características nucleares similares a las papilares" (NIFTP, por sus siglas en inglés), anteriormente conocida como “PTC no invasiva con variante folicular” (FVPTC, por sus siglas en inglés), fue rebautizada como NIFTP para resaltar su comportamiento indolente y su tasa de recurrencia menor del 1%. Estas neoplasias se consideran benignas, pero tienen potencial maligno, y se requiere extirpación quirúrgica para confirmar que no son invasivas.
- Cáncer folicular de tiroides (FTC): El carcinoma folicular representa del 8-9% de los cánceres de tiroides pediátricos. - Cáncer medular de tiroides (MTC): El carcinoma medular representa el 4% de los cánceres de tiroides pediátricos, pero una mayor proporción en niños pequeños. La mayoría de los casos se asocian a neoplasia endocrina múltiple tipo 2 (NEM2).
Las tasas de supervivencia a largo plazo para PTC o FTC se aproximan al 98%. Las tasas de supervivencia a cinco años para el CMT también superan el 90%.
MANIFESTACIONES CLÍNICAS:
Tanto el cáncer de tiroides diferenciado (CDT) como el cáncer medular de tiroides (MTC) en niños suelen presentarse como nódulos solitarios asintomáticos. La gran mayoría de los niños con nódulos tienen una función tiroidea normal. Una minoría de niños con un nódulo tiroideo presenta características clínicas y de laboratorio de hipertiroidismo debido a un nódulo autónomo ("adenoma tóxico"), que es relativamente raro en los niños.
- CDT: incluye el cáncer papilar de tiroides (PTC) y el cáncer folicular de tiroides (FTC). Los hallazgos clínicos que aumentan la probabilidad de cáncer en un niño que presenta un nódulo tiroideo son el sexo masculino, los antecedentes de radiación externa a la cabeza y el cuello, los antecedentes de crecimiento rápido del nódulo, una masa firme o fija, ronquera o disfagia y adenopatía cervical.
- MTC: Suelen presentar un nódulo solitario o se descubren de manera incidental cuando un miembro de la familia es diagnosticado con MTC, generalmente como parte de una neoplasia endocrina múltiple tipo 2A (NEM2A) o NEM2B. Los niños con NEM2B pueden tener un Síndrome de habitus corporal marfanoide, neuromas mucosos de la lengua y las conjuntivas, y nervios corneales medulares (engrosados).
DIAGNÓSTICO:
Para identificar a los pacientes de cáncer se usa un proceso diagnóstico mediante pruebas de función tiroidea, gammagrafía tiroidea, ecografía y punción aspiración con aguja fina (PAAF).
La PAAF es la prueba más útil para diferenciar los nódulos tiroideos benignos del cáncer y se prefiere a la biopsia con aguja gruesa, que es más invasiva y tiene un mayor riesgo de complicaciones. Además, tiene una alta precisión diagnóstica en niños.
TRATAMIENTO:
Cáncer papilar de tiroides:
El PTC representa más del 80% del cáncer de tiroides en niños y es la forma más común de cáncer de tiroides diferenciado (CDT). Para la mayoría de los niños con PTC que se diagnostica o se sospecha fuertemente en función de la evaluación preoperatoria, sugerimos una tiroidectomía total o casi total en lugar de una lobectomía.
La tiroidectomía total ha sido durante mucho tiempo el procedimiento quirúrgico inicial recomendado porque la PTC en los niños es bilateral en el 35 al 45% de los casos y esta característica puede no ser detectable en las imágenes preoperatorias. Además, la resección bilateral de la tiroides se ha relacionado con tasas más bajas de recurrencia de la enfermedad en comparación con la resección unilateral en algunos estudios.
Cáncer folicular de tiroides:
El FTC representa aproximadamente del 8 al 9% de los CDT en los niños. A diferencia de la PTC, la FTC tiende a ser unifocal, por lo general, se presenta en nódulos con citología por PAAF.
Para los pacientes con hallazgos de PAAF relacionados con una neoplasia folicular para quienes se recomienda cirugía, el primer paso generalmente es una lobectomía diagnóstica. En el caso de la FTC mínimamente invasiva, la lobectomía suele ser curativa.
Cáncer medular de tiroides:
La tiroidectomía total es el tratamiento de elección para los niños con cáncer medular de tiroides (MTC).
Para los pacientes pediátricos sin MTC, pero con un alto riesgo de desarrollar MTC debido a mutaciones en el gen RET, se recomienda la tiroidectomía total "profiláctica" durante la infancia o la primera infancia. En los pacientes con mutaciones de riesgo más alto (neoplasia endocrina múltiple tipo 2B ‹NEM2B›), se recomienda la tiroidectomía durante el primer año de vida.
Tumores de Ovario
Los tumores ováricos son raros en la edad pediátrica y representan del 1 al 5% de los tumores infantiles.
La presentación en la edad pediátrica es bimodal, una entre los 2-3 años y otra entre los 12-15 años. El pico máximo de incidencia aparece en la adolescencia, con más del 50% de los casos, siendo extremadamente raros por debajo del año de vida.
De acuerdo con la clasificación de la OMS, se divide en tres grupos: tumores del epitelio de superficie, tumores de células germinales y tumores del estroma y cordones sexuales.
En la edad pediátrica, los tumores derivados de células germinales representan hasta el 90% de los tumores ováricos, siendo el tipo histológico más frecuente el teratoma quístico maduro o tumor dermoide.
Si excluimos las tumoraciones químicas benignas, los tumores sólidos predominantes en el aparato genital femenino en niñas y adolescentes son los tumores germinales, que representan hasta el 90%, una diferencia de la población femenina adulta, donde predominan los tumores derivados del epitelio, un 75% del total, siendo el 80% carcinomas, en comparación con los derivados de células germinales, que suponen el 15-25%.
La clínica de los tumores de ovario es bastante inespecífica; predomina el dolor, la distensión abdominal y la palpación de una masa. El dolor abdominal suele ser insidioso, acompañado de distensión abdominal, pero puede ser de inicio agudo debido a torsión, rotura o hemorragia, comenzando como un abdomen agudo quirúrgico.
El diagnóstico, al igual que en la población adulta, se basa en las pruebas de imagen, los marcadores tumorales y el estudio anatomopatológico.
La ecografía ayuda a la localización de la masa y a definir sus características. Los tumores malignos suelen tener bordes irregulares y necrosis en el centro de la tumoración, y aparecen como masas de tejido blando mal definidas. En cambio, los tumores benignos son hipoecoicos y pueden presentar un refuerzo acústico posterior. Los quistes ováricos son generalmente anecoicos y de paredes finas.
La TAC es considerada hasta hoy día el estándar de oro para el estudio de extensión. La RMN se reserva para aquellos casos en los que, debido al gran tamaño de la tumoración, haya dudas sobre el origen de la masa.
Los marcadores tumorales, si bien no confirman el diagnóstico de benignidad o malignidad de la tumoración (ya que hasta en un 20% de los casos son positivos en tumores benignos), son de gran ayuda para la orientación terapéutica, el seguimiento posterior y la respuesta al tratamiento realizado. Los más utilizados son la alfa fetoproteína (AFP), la subunidad beta de la gonadotropina coriónica humana (BHCG) y la lactato deshidrogenasa (LDH).
El diagnóstico de confirmación es el estudio anatomo patológico, ya sea tras la resección quirúrgica o cuando esté indicado a través de toma de biopsia con aguja fina guiada por ecografía.
En pediatría, los tumores malignos de origen ovárico son minoría y la clínica es inespecífica; por tanto, no podemos dejar que pasen desapercibidos y hay que hacer un diagnóstico precoz para tener una actitud terapéutica adecuada.
El tratamiento que se debe adoptar dependerá del tipo de tumor y de su extensión. La cirugía juega un papel fundamental en el tratamiento de los tumores ováricos, y puede extirpar la masa completa en más del 90%.
Es indispensable seguir los principios estrictos de la cirugía oncológica, realizando en la mayoría de los pacientes salpingo-ooforectomía mediante laparotomía o por abordaje laparoscópico. Cuando en la evaluación preoperatoria no se pueda asegurar la resecabilidad del tumor maligno, procede practicar biopsia por punción ecoguiada y, tras la quimioterapia reductora, programar la exéresis tumoral.
Tradicionalmente se ha defendido que el uso de la laparoscopia para el tratamiento de lesiones malignas o altamente sospechosas no estaba aconsejado. Actualmente, con el desarrollo de la cirugía mínimamente invasiva, esto ha dejado de ser válido y el abordaje depende de las características de la masa. En los tumores benignos y en los bilaterales se intenta realizar una cirugía conservadora con preservación del lado menos afectado, para intentar conservar la fertilidad de la paciente a largo plazo.
Tumores de Testículo
Los tumores testiculares son infrecuentes en niños y representan el 1-2% de las neoplasias sólidas, con presentación bimodal: el primero entre los 2 y los 4 años de edad y el segundo en torno a los 15 años.
En la exploración física, encontraremos una masa delimitada o bien el teste globalmente aumentado de tamaño, con una consistencia pétrea y transiluminación concordante con masa sólida.
Los tumores testiculares se dividen en 7 grupos (clasificación anatomo-patológica según la Organización Mundial de la Salud de 2016):
- Tumores de células germinales derivados de neoplasia de células germinales in situ (NCGIS) que incluyen seminoma, carcinoma embrionario, tumor del saco vitelino pospuberal, teratoma de tipo pospuberal, tumores trofoblásticos y tumores de células germinales de tipo desconocido.
- Tumores de células germinales no relacionados con NCGIS que comprenden tumor espermatocítico, teratoma de tipo prepuberal, tumor del saco vitelino de tipo prepuberal y tumor mixto teratoma-tumor del saco vitelino de tipo prepuberal.
- Tumores de los cordones sexuales/estroma gonadal (tumores de células de Leydig, de Sertoli o de la granulosa).
- Tumores que contienen elementos de células germinales y de cordones sexuales/estroma gonadal.
- Tumores misceláneos.
- Tumores hemato-linfoides.
- Tumores de los conductos y la rete testis.
Los tumores malignos se diseminan por vía linfática y hematógena. La diseminación por vía linfática a los ganglios retroperitoneales solo ocurre en el 4-6%. Aparecen metástasis por vía hematógena en el 9% de los niños (frente al 60% de los adultos), el asiento más frecuente de estos es el pulmón. El pronóstico global del tumor testicular prepuberal es muy bueno, con una supervivencia a los 5 años que se acerca al 99%.
La orquiectomía radical se ha considerado como el tratamiento estándar de masas testiculares en todas las edades. Sin embargo, en los niños, debido a su comportamiento benigno, en los últimos años se ha replantado la actitud médico-quirúrgica. Esto supone un cambio en el abordaje terapéutico inicial, al tomar en consideración la realización de una cirugía conservadora (tumorectomía), basados en los valores de los marcadores tumorales (B-HCG, AFP), tamaño tumoral y hallazgos histopatológicos.
Retinoblastoma
El 90% se manifiesta antes de los 3 años y puede estar presente al nacimiento. 2/3 de los casos son unilaterales y 1/3 bilaterales. El tumor resulta de dos mutaciones del cromosoma 13, que codifica una proteína reguladora de la proliferación celular, por lo que habrá un crecimiento descontrolado.
El signo más frecuente es la leucocoria, seguido del estrabismo, pero puede simular una celulitis preseptal, manifestarse como glaucoma doloroso o cambios en el color del iris.
Fuente: Sant Joan de Déu. Retinoblastoma pediátrico. 2019.
La tasa de supervivencia en nuestro medio es mayor del 90%, donde en 2/3 el ojo se conservará, mientras que en países en vías de desarrollo la supervivencia es tan solo del 45%. El tratamiento de elección es la quimioterapia intraarterial en la arteria oftálmica. Otros tratamientos son: la quimioterapia sistémica e intravítrea, braquiterapia, crioterapia, termoterapia y la enucleación, que consisten en la extracción del globo ocular. Dependerán del estadio de la enfermedad y del estado del paciente.
3.5 PROCESO DE ATENCIÓN DE ENFERMERÍA: NANDA-NOC-NIC
A continuación, se muestran unos Diagnósticos de Enfermería clasificados dentro de los Patrones Funcionales de Marjory Gordon. Estos NANDA se asocian a una serie de Resultados (NOC) y de Intervenciones (NIC) a modo de conceptualización, debido a que un Proceso de Atención de Enfermería es dinámico y, además, evoluciona a lo largo del tiempo, según cambia la situación del paciente en el proceso de salud-enfermedad. De esta forma, debemos determinar las intervenciones a realizar en cada una de sus etapas, de forma prioritaria, para satisfacer las necesidades alteradas.
PATRÓN 1-Percepción-Manejo de la salud:
PATRÓN 2-Nutricional-Metabólico:
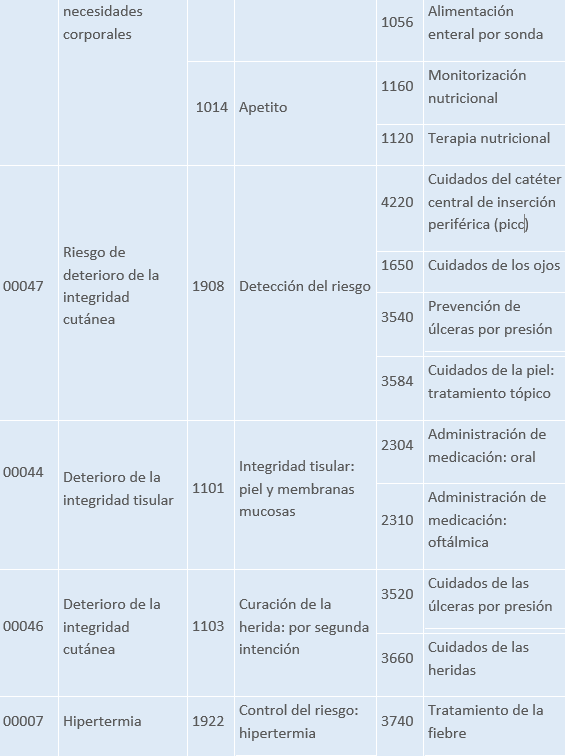
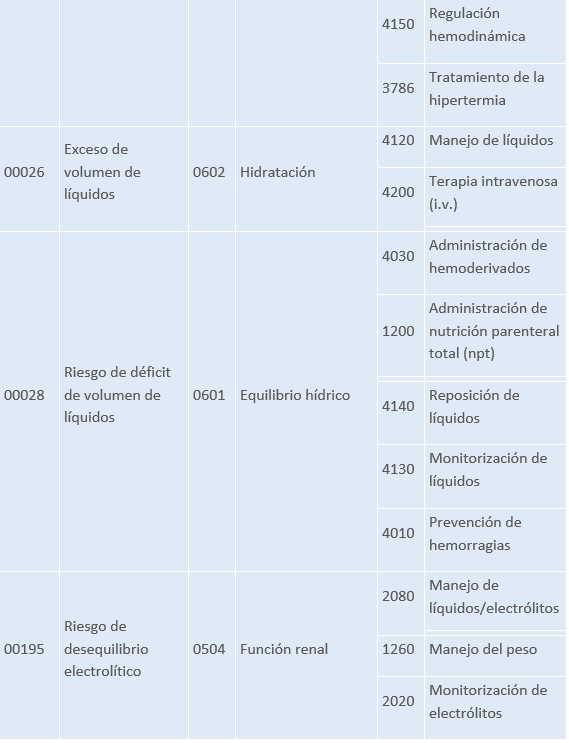
PATRÓN 3-Eliminación:
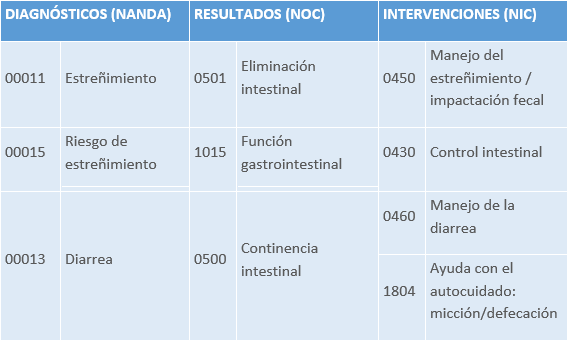
PATRÓN 4-Actividad-Ejercicio:
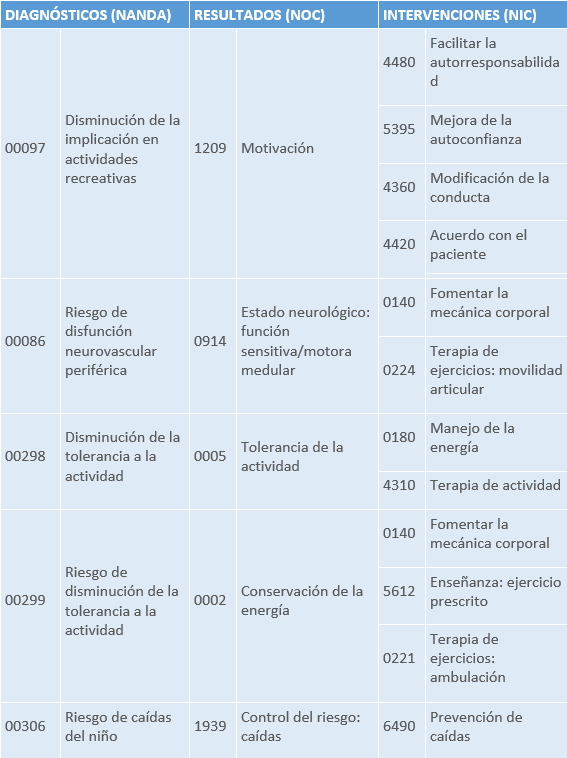
PATRÓN 5-Sueño-reposo:
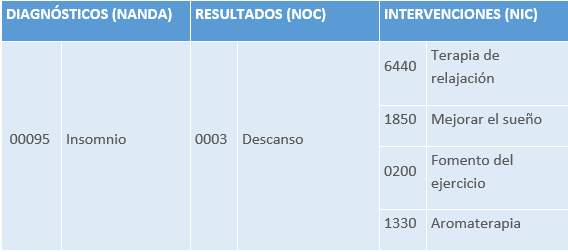
PATRÓN 6-Cognitivo-perceptivo:
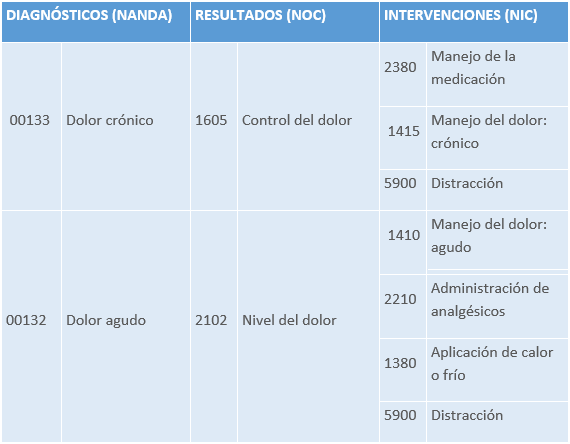
PATRÓN 7-Autopercepción-autoconcepto:
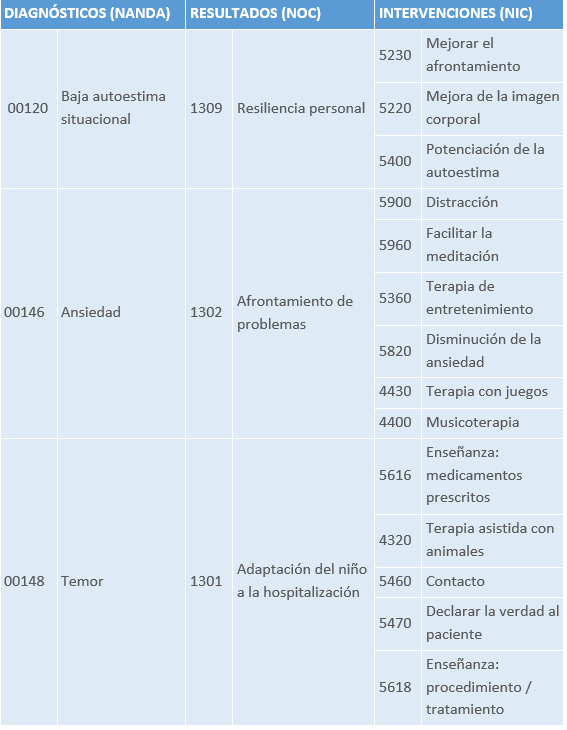
PATRÓN 8-Rol-Relaciones:
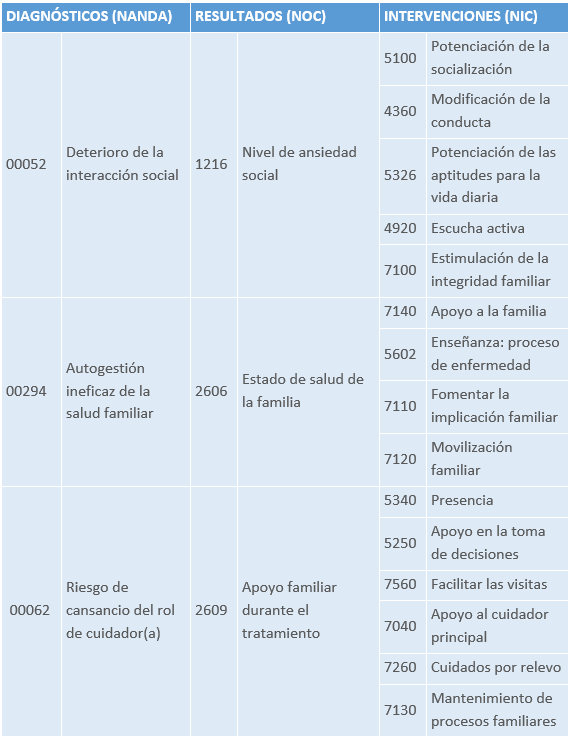
PATRÓN 9- Sexualidad-Reproducción.

PATRÓN 10-Afrontamiento-Tolerancia del estrés:
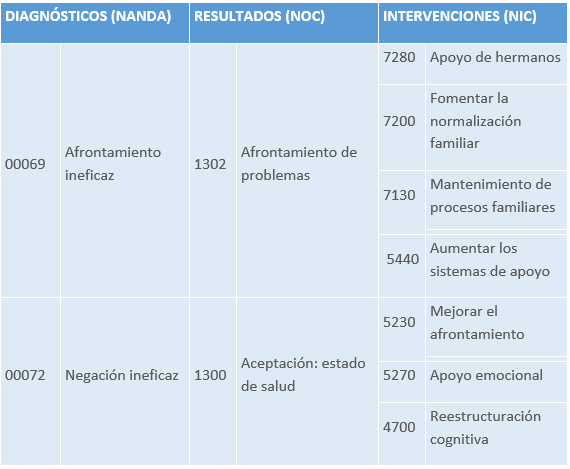
3.6 CUIDADOS EN NEFROUROLOGÍA: PROCEDIMIENTOS Y TÉCNICAS
3.6.1 Tratamiento del cáncer
Cirugía
Antes de la aplicación de la quimioterapia y radioterapia, la cirugía constituía el único tratamiento posible de los tumores sólidos; no obstante, solo un pequeño porcentaje de pacientes con tumores localizados se curaban. Actualmente, es conocido que el abordaje del cáncer infantil requiere un tratamiento multimodal, siendo la cirugía una parte importante de este.
El papel tradicional de la cirugía en el tratamiento del cáncer, ha sido el de la extirpación tumoral. Sin embargo, en ocasiones, nos encontramos con tumores irresecables por compromiso de estructuras vitales, es en estos casos cuando el niño puede beneficiarse de la reducción de la masa tumoral mediante resecciones parciales o casi totales, para posteriormente administrar radioterapia y/o quimioterapia; es decir, practicando una citorreducción del tumor. La resección de las metástasis en el cáncer infantil se suele realizar con un fin curativo y no suelen abordarse hasta no haber resecado el tumor primario. Los tumores que con más frecuencia requieren la resección de metástasis son: el ostosarcoma, el sarcoma de Ewing, el tumor de Wilms, el hepatoblastoma y los tumores de partes blandas. En muchos de estos tumores, las metástasis suelen ser pulmonares.
Las indicaciones quirúrgicas varían dependiendo del tipo de tumor. A continuación, se exponen las peculiaridades del tratamiento quirúrgico de algunos de los tumores en Pediatría:
- El tratamiento quirúrgico del tumor de Wilms es controvertido, los dos principales grupos cooperativos recomiendan esquemas de tratamiento opuestos, la Sociedad Europea de Oncología Pediátrica recomienda administrar quimioterapia neoadyuvante y, posteriormente, realizar la resección quirúrgica del tumor, con el fin de reducir el tamaño tumoral por un lado y, por otro, la posibilidad de complicaciones asociadas a la cirugía, como la hemorragia o la diseminación tumoral. El Grupo de Estudio del Tumor de Wilms propone realizar una nefrectomía radical inicial sin quimioterapia neoadyuvante previa, alegando la necesidad de obtener un diagnóstico histopatológico y correcto estadiaje del tumor desde el principio.
- En el neuroblastoma, el papel de la cirugía es variable, en función de la localización y extensión del tumor. Los tumores en estadios precoces y con marcadores biológicos favorables se benefician de la resección quirúrgica sin asociar otras modalidades de tratamiento. En el resto de tumores, el tamaño tumoral o la localización hacen que la resección completa inicial sea poco probable o muy compleja quirúrgicamente, por lo que precisan de quimioterapia neoadyuvante.
- Los rabdomiosarcomas son tumores de origen mesenquimal que se localizan con mayor frecuencia en el cuello (35%), el tracto genitourinario (25%) y las extremidades (19%). Suelen extirpar aquellos cuya extensión es limitada y la localización accesible quirúrgicamente; en el resto de los pacientes, la quimioterapia y la radioterapia son el tratamiento de elección.
- El papel de la cirugía en el tratamiento de los linfomas, como los linfomas de Hodgkin, se limita, en la mayoría de los casos, a establecer el diagnóstico mediante la biopsia. Con la mejora de las técnicas de diagnóstico por imagen, la mayoría de los protocolos contemplan su estadiaje clínico y no quirúrgico.
Quimioterapia
Formas de quimioterapia:
- Quimioterapia de inducción. Se emplea como tratamiento primario en pacientes con enfermedad avanzada o diseminada, o en aquellos pacientes en los que no existe otra posibilidad de tratamiento. Ofrece ventajas: el tratamiento actúa sobre todas las posibles localizaciones tumorales incluidas las microscópicas y permite un tratamiento más agresivo.
- Quimioterapia adyuvante. Consiste en administrar quimioterapia sistémica tras el control local del tumor primario mediante cirugía y/o radioterapia en pacientes con alto riesgo de recaída metastásica, como los que presentan tumores de gran volumen. La finalidad de la quimioterapia adyuvante es eliminar la enfermedad microscópica presente al diagnóstico y que no se elimina con el tratamiento inicial. Se ha demostrado eficaz en algunos tumores pediátricos frecuentes, incluyendo: el tumor de Wilms, los linfomas, el sarcoma de Ewing, el osteosarcoma y el rabdomiosarcoma.
- Quimioterapia neoadyuvante. Consiste en el empleo de la quimioterapia como tratamiento inicial en los pacientes con cáncer localmente avanzado, aunque todavía limitado. Reduce el tamaño del tumor y facilita el tratamiento quirúrgico/radioterápico posterior, también disminuye las posibilidades de complicaciones derivadas de estos procedimientos, como hemorragias o diseminación tumoral intraoperatoria. Además, permite valorar la respuesta histológica del tumor al tratamiento inicial con citostáticos y proporciona tratamiento precoz de la enfermedad microscópica metastásica.
- Quimioterapia de rescate. El fracaso de los protocolos de terapia combinada reduce las posibilidades de éxito en el tratamiento del tumor. En caso de reincidencia de la enfermedad, se debe emplear una segunda línea de tratamiento compuesta por fármacos con diferentes mecanismos de acción a los empleados con anterioridad. Este tipo de tratamiento tiene como objetivo, obtener una nueva remisión completa de la enfermedad. Los resultados terapéuticos de este tipo de tratamiento son peores, de modo general, a los obtenidos con los esquemas iniciales.
Resistencia a fármacos:
El principal motivo de fracaso terapéutico en el cáncer infantil es la resistencia a citostáticos. Los mecanismos de quimiorresistencia tienen un papel fundamental a la hora de seleccionar los fármacos que se incluyen en los distintos regímenes de quimioterapia y en los tratamientos de segunda línea (quimioterapia de rescate). Esta resistencia puede existir desde el inicio del tratamiento en tumores expuestos a quimioterapia por primera vez o bien puede adquirirse a lo largo del tratamiento. También, puede ser específico para un solo agente o inespecífica, afectando entonces de forma simultánea a distintas clases de antineoplásicos, es lo que se conoce como resistencia a múltiples drogas.
Radioterapia
La radioterapia constituye un procedimiento terapéutico de gran eficacia en todos los grupos de edad. No obstante, su aplicación en la infancia requiere una atención especial, debido a sus potenciales efectos sobre los tejidos en fase de crecimiento y desarrollo y el riesgo de segundos tumores a largo plazo. En los últimos años, se han producido importantes avances tecnológicos en el empleo de esta modalidad terapéutica, que permiten irradiar de forma precisa el tumor, minimizando los efectos secundarios.
La radioterapia consiste en depositar una determinada cantidad de energía en un área específica de tejido previamente seleccionado, con el objetivo de destruir las células malignas. Produce daños en el ADN de las células, tanto sanas como malignas, mediante una ionización de sus átomos. Las células sanas, en general, son capaces de reparar estos daños con más facilidad, mientras que las células tumorales tienen alterados los mecanismos de reparación normales y resultan más afectadas por el tratamiento.
Las bases biológicas de la radioterapia se fundamentan en las diferentes respuestas del tejido sano y tumoral a las radiaciones ionizantes, en función de la dosis y los tiempos de administración. La dosis total de radiación se administra con frecuencia de forma fraccionada en distintas sesiones, durante un período de tiempo variable que oscila entre 3-6 semanas en la mayoría de los casos, esto se conoce como fraccionamiento de la dosis.
El fraccionamiento de la dosis total de radiación permite a las células normales reparar los daños subletales ocasionados por la radiación sin permitir que lo hagan las tumorales. El acumulado de lesiones subletales en la célula neoplásica provoca un daño letal en esta. La relación tiempo/dosis de la radioterapia se define por: la dosis total de radiación (medida en Grays), el número de “fracciones” necesarias para administrar la dosis total, la dosis por fracción y la duración del tratamiento.
Las dosis de radiación empleadas y la forma de administración dependen de la edad del paciente, la localización, el tipo y la radiosensibilidad del tumor.
- Esquemas de fraccionamiento:
En la mayoría de los centros, el fraccionamiento convencional consiste en administrar entre 1,8 y 2 Grays (Gy) por fracción, una vez al día, durante 5 días a la semana, hasta completar dosis totales de radiación entre 50-60 Gy.
En niños, tanto la dosis por fracción como la dosis total de radiación, se ajustan de forma individualizada al tipo de tumor. Tumores muy radiosensibles, como los linfomas de Hodgkin, la leucemia aguda y los germinomas reciben dosis mínimas por fracción de 1,5 Gy.
- Modalidades de radioterapia:
- Radioterapia con haz externo:
La forma más habitual de administrar la radiación es a través de una fuente localizada externamente al paciente. Para ello, se emplean aceleradores capaces de generar haces de fotografías, electrones o partículas pesadas. Los fotones tienen más penetrancia y capacidad de alcanzar órganos internos y los electrones se usan habitualmente para objetivos más superficiales.
La radioterapia con haces de protones es de gran interés en el tratamiento del cáncer en niños, principalmente en los tumores del sistema nervioso central. Los protones son partículas pesadas que no eliminan su energía según van penetrando en los tejidos como ocurre con los fotones, su máximo pico de radiación se libera cuando se para en el interior de los tejidos, propiedad que permite calcular la energía del haz de protones y cuál será su trayectoria. Está técnica reduce de forma significativa la dispersión de la radiación en los tejidos sanos.
Uno de los grandes avances en la radioterapia oncológica se produce en los años 80 con la introducción de la tomografía computarizada (TC), que permite realizar reconstrucciones tridimensionales del área clínica a irradiar. Con la radioterapia de conformación tridimensional (TC3D) se obtienen imágenes con TC o resonancia magnética nuclear (RMN) que permiten delimitar de forma precisa el área del tumor y las zonas de diseminación microscópica, evitando estructuras vitales y tejidos normales.
Otra modalidad de radioterapia introducida en los últimos años, ha sido la radioterapia de intensidad modulada (IMRT), que utiliza múltiples campos de radiación y modifica la intensidad de los haces de colimación localmente durante la radiación, a diferencia de la radiación convencional en la que el haz de radiación es uniforme a lo largo del campo. Esta técnica presenta, sin embargo, un inconveniente, el riesgo elevado de no irradiar zonas de enfermedad microscópica debido a la gran definición que posee. Este problema se intenta obviar mediante las técnicas de imagen funcional, como la tomografía de emisión de positrones (PET) o la resonancia magnética espectroscópica (RMS). Con estas técnicas, pueden incluso crearse zonas cóncavas de distribución de la dosis, lo cual es muy útil en determinadas localizaciones, como en algunos tumores de tejidos blandos o para evitar radiar la médula espinal o la cabeza del húmero. Su papel también es fundamental, cuando queremos evitar sobrerradiar determinadas estructuras de importancia próximas al tumor, como la cóclea en el tratamiento de los tumores de fosa posterior. Con las nuevas tecnologías, se ha conseguido reducir hasta un 65% las dosis de radiación sobre la cóclea y, consecuentemente, disminuir la ototoxicidad en niños.
La radioterapia fraccionada estereotáctica constituye otra modalidad de radioterapia de reciente uso en Pediatría.
- Braquiterapia:
Consiste en colocar sustancias radioactivas en contacto directo con el tejido tumoral, con el fin de depositar dosis muy elevadas de radiación en el tumor. En la población infantil, la braquiterapia puede presentar ventajas sobre la radiación externa en determinados tumores, como: rabdomiosarcomas pélvicos (uterinos, vaginales o perirrectales), retinoblastomas (cuando el tumor es de pequeño tamaño, unilateral y con mínimo riesgo de enfermedad multifocal) o en sarcomas de tejidos blandos de las extremidades inferiores, reduciendo las dosis de radiación en los cartílagos de crecimiento. Su uso no está muy extendido, debido a que, desde el punto de vista práctico, la braquiterapia se limita a lechos tumorales cuyo diámetro mayor no es superior a los 5-10 cm.
Otras aplicaciones de la radioterapia:
En algunos protocolos de acondicionamiento previo al trasplante alogénico de progenitores hematopoyéticos, se combina de forma secuencial la irradiación corporal total (12 Gy en 6 fracciones) con las altas dosis de quimioterapia.
La radioterapia también se usa con fines paliativos en los tumores infantiles. Se ha demostrado eficaz en el tratamiento del dolor que no responde a analgésicos u otros procedimientos. Las pautas de tratamiento son variables en función de la expectativa de vida del paciente, desde una sola sesión hasta tratamientos durante varias semanas.
Por último, no hay que olvidar la aplicación de la radioterapia en situaciones de urgencia oncológica, como los síndromes de compresión medular o el síndrome de vena cava superior, entre otros.
- Toxicidad de la radioterapia:
La toxicidad de la radioterapia sobre los tejidos normales puede observarse durante el tratamiento o años después. Según el momento de aparición, los efectos adversos se pueden agrupar en:
- Agudos: Aquellos que tienen lugar durante o inmediatamente después de la radiación y que pueden modificarse con la intensidad de la dosis.
- Subagudos: Los efectos adversos que tienen lugar entre los 3 y 9 meses del tratamiento.
- Tardíos: los que aparecen de forma característica al año de recibir la radioterapia.
La toxicidad de la radiación, depende de factores, como: el área anatómica irradiada, los parámetros del tratamiento (dosis de radiación total, esquemas de fraccionamiento y dosis por fracción), factores intrínsecos del huésped (estado nutricional, hipoxemia y factores genéticos celulares que predisponen al daño por radiación) y de la asociación de otros tratamientos de forma simultánea (cirugía y/o quimioterapia).
- Toxicidad aguda:
Los efectos secundarios agudos son considerados normales y esperables. Aparecen en tejidos con un elevado índice de proliferación celular, como: los epitelios, el aparato digestivo y las células hematopoyéticas. Los cambios en los epitelios pueden objetivarse de forma inmediata tras la exposición a la radiación.
El eritema cutáneo y la sialoadenitis se manifiestan a las pocas horas y son más frecuentes en la radiación corporal total que en la radioterapia local. La alopecia aparece en la tercera semana de la radiación craneal y la recuperación parcial o total del pelo se produce a los 3 meses del tratamiento, dependiendo de las dosis de radiación que recibe la zona tratada.
La afectación de las mucosas presenta características similares a las lesiones de la piel, se manifiesta como un intenso enantema que, en ocasiones, se complica al sobreinfectarse por cándida. Cuando las lesiones se extienden a la hipofaringe y esófago, el niño puede presentar distintos grados de odinofagia.
Los efectos secundarios sistémicos (náuseas, vómitos, astenia y anorexia) ocurren a los días de iniciar el tratamiento, y su frecuencia e intensidad dependen de la superficie y localización en la zona irradiada.
La afectación de la hematopoyesis es variable, la radiación local de la médula ósea provoca una linfopenia precoz. Al final de la segunda semana de tratamiento, descienden los recuentos de neutrófilos de forma poco significativa, en los procedimientos con superficies radiadas de gran tamaño (radiación cráneo-espinal, torácica o abdominal extensa). En aquellos procedimientos en los que se irradia la médula ósea (pelvis), las cifras de neutrófilos disminuyen de forma significativa; de forma similar las plaquetas bajan, también, entre la segunda y tercera semana de tratamiento.
La quimioterapia asociada puede aumentar, acelerar o retrasar la aparición de toxicidad aguda en los tejidos normales, especialmente cuando se administra actinomicina D, metotrexato o cisplatino. El cisplatino a bajas dosis, administrado junto con la radiación, produce un efecto sinérgico conocido como radiosensibilización. Por el contrario, la radioterapia también puede alterar el metabolismo de los citostáticos aumentando su toxicidad, un ejemplo sería el de la radiación craneal, que asociada a la administración de metotrexato sistémico puede producir un cuadro de leucoencefalopatía necrotizante en los pacientes pediátricos.
- La toxicidad subaguda y tardía:
Se manifiesta después de 3 meses de la radiación. Los efectos a largo plazo pueden aparecer hasta años después del tratamiento.
Estos efectos tardíos son irreversibles y progresivos, y se deben principalmente a la lesión del endotelio vascular y de las células parenquimatosas del tejido radiado. Incluyen: cuadros de fibrosis pulmonar intersticial progresiva, pericarditis, disfunción tiroidea e hipofisaria, nefritis y obstrucción intestinal, entre otros.
Sin embargo, uno de los principales inconvenientes de la radioterapia en la infancia es su efecto sobre los tejidos en crecimiento y desarrollo. Se han objetivado secuelas neurológicas y cognitivas en los niños con tumores del sistema nervioso central irradiados a edades tempranas. La toxicidad subaguda sobre el sistema nervioso central produce la destrucción progresiva de la sustancia blanca junto con cambios en los astrocitos responsables del mantenimiento de la barrera hematoencefálica. Clínicamente, la toxicidad subaguda de la radioterapia craneal se traduce en el denominado “síndrome de somnolencia” (somnolencia, anorexia y febrícula) que prácticamente aparece a las 4-8 semanas de la radiación en la leucemia aguda linfoblástica o en los tumores primarios del SNC.
De forma paralela, durante las 4-8 semanas (aunque puede prolongarse hasta 12 meses), la radiación puede empeorar de forma transitoria los síntomas propios de la enfermedad como consecuencia de la necrosis tumoral que produce el tratamiento, los síntomas responden al tratamiento con esteroides.
Los efectos tardíos sobre el SNC son secundarios a la necrosis focal que aparece a los 6-24 meses de la radiación, se producen cuando la dosis total supera los 60Gy o en dosis por fracción superiores a los 2,4-3 Gy y la probabilidad de estas lesiones es mayor en niños menores de 5 años.
Los efectos neurocognitivos provocados por la radioterapia están claramente descritos en la literatura. La magnitud del grado de afectación del coeficiente intelectual (CI) depende de la dosis y de la edad del niño, y suele manifestarse a los 2-5 años de la radioterapia. El descenso del CI es secundario al déficit de atención y las alteraciones en los procesos de memoria, pudiendo contribuir, también, los déficits neurológicos tras la neurocirugía. Este descenso del CI afecta principalmente a los niños entre 3-5 años, por lo que muchos protocolos de tratamiento no incluyen la radiación craneal en niños menores de esta edad. También, se ha comprobado un efecto sobre el crecimiento óseo por alteraciones de los cartílagos epifisarios. La magnitud de dicho retraso dependerá de la edad en el momento de la radiación y de la dosis administrada; En general, dosis superiores a 20 Gy tienen un efecto significativo en el crecimiento óseo.
Finalmente, el desarrollo de tumores secundarios está documentado hasta en un 5,8% de los supervivientes de cánceres infantiles que recibieron radioterapia durante el tratamiento de su neoplasia. Los tumores secundarios a radiación más frecuentes son: el cáncer de mama, los sarcomas de tejidos blandos, como el osteosarcoma, y los carcinomas de piel y tiroides, que se desarrollan entre 9 y 16 años después de la radiación.
3.6.2 Complicaciones
Neutropenia febril
A pesar de los avances en el tratamiento y el aumento de la supervivencia en el cáncer infantil, las infecciones continúan siendo una causa importante de morbimortalidad, especialmente en los pacientes que reciben quimioterapia y asocian neutropenia, pudiendo ser la fiebre el único signo presente o asociarse a signos y síntomas inespecíficos.
Los pacientes pediátricos que reciben quimioterapia suelen presentar una alteración de las barreras mucosas (mucositis) que predispone a la infección por microorganismos oportunistas de la microbiota de la piel, de la mucosa oral y del tracto gastrointestinal. Por otra parte, el aumento de las resistencias a antimicrobianos en las infecciones relacionadas con la asistencia sanitaria complica el abordaje de estos pacientes.
La definición de neutropenia febril (NF) implica la coexistencia de:
- Neutropenia: recuento absoluto de neutrófilos ≤500mm3 o ≤1.000mm3 con expectativa de descenso en las siguientes 24-48h por debajo de 500mm3.
- Fiebre: registro único de temperatura axilar ≥38,3-38,5°C (>38°C en las recomendaciones del Reino Unido y Australia) o 2 mediciones ≥38°C separadas/mantenidas al menos una hora.
Se aísla un microorganismo en hemocultivo entre el 10-30% de los episodios febriles en pacientes pediátricos con NF. Se ha evidenciado un incremento progresivo en la prevalencia de bacilos gramnegativos multirresistentes.
La valoración inicial del paciente pediátrico oncohematológico con NF debe incluir una exhaustiva anamnesis general y dirigida a detectar posibles focos infecciosos, así como a valorar el grado de inmunosupresión y riesgos asociados, una cuidadosa exploración física completa y la realización de analítica sanguínea con hemograma, bioquímica y biomarcadores de respuesta inflamatoria, como la proteína C reactiva (PCR) y la procalcitonina (PCT).
La realización de otras pruebas complementarias, que no deben retrasar el inicio del tratamiento, incluye la recogida de hemocultivos de todas las luces de los catéteres centrales. Además, hay que valorar la extracción de hemocultivo venoso periférico, especialmente si sospecha de infección relacionada con el catéter vascular central (extraídos en forma de hemocultivos cuantitativos o cualitativos diferenciales en tal caso, asegurando el mismo volumen y momento de incubación), muestra de orina no invasiva y detección de virus respiratorios si periodo epidémico y/o síntomas respiratorios. Otras pruebas complementarias estarán dirigidas por signos o síntomas.
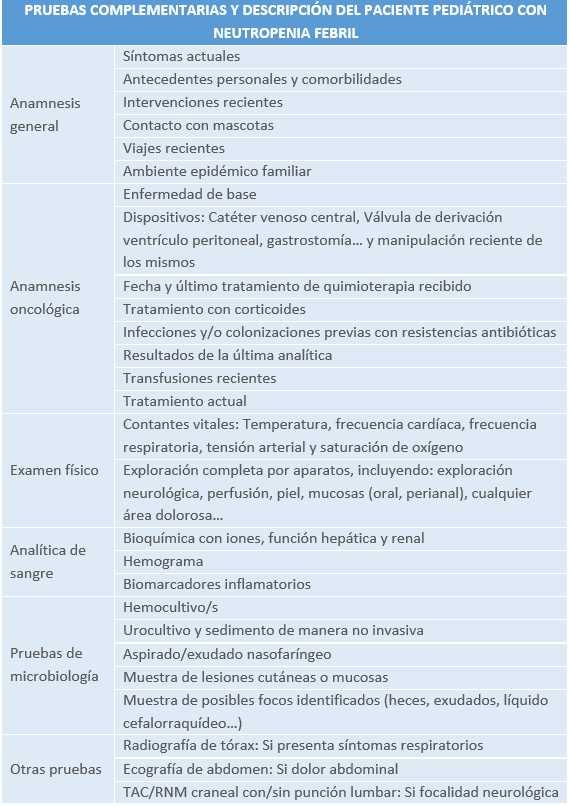
Terapia antibacteriana recomendada:
El inicio precoz del tratamiento antibiótico de amplio espectro en estos pacientes es un aspecto fundamental que condiciona el pronóstico. Debido al elevado riesgo de infecciones por P. aeruginosa y su alta morbimortalidad, se recomienda su cobertura empírica.
- Pacientes de alto riesgo:
- Pacientes hemodinámicamente estables:
La terapia combinada no ha demostrado mejores resultados clínicos en pacientes estables. Por ello, el tratamiento combinado de un β-lactámico junto a un segundo antibiótico quedará reservado para situaciones específicas, como shock séptico y/o riesgo de microorganismos multirresistentes, teniendo en cuenta la epidemiología de cada centro.
En el caso de pacientes con un foco infeccioso identificado se seleccionará la antibioterapia empírica según los microorganismos más frecuentes en cada foco, asegurando el buen control del mismo (p. ej., retirada de catéteres infectados, drenaje de abscesos, etc.).
- Pacientes hemodinámicamente inestables:
Los pacientes oncológicos con shock séptico presentan una mortalidad elevada. Entre los principales factores asociados a la mortalidad se encuentra el uso de tratamiento antibiótico empírico inadecuado, sin actividad frente al agente causal. Por ello, se recomienda ampliar el espectro antibiótico con el objetivo de aumentar las posibilidades de incluir un agente activo frente al microorganismo implicado, considerando su ajuste posterior según la evolución y aislamientos microbiológicos.
Los nuevos antibióticos comercializados (p. ej., ceftazidima-avibactam, ceftolozano-tazobactam, meropenem-vaborbactam, cefiderocol) ofrecen nuevas oportunidades de tratamiento. Su uso de forma empírica será restringido a pacientes hemodinámicamente inestables con infecciones o colonizaciones recientes resistentes a los carbapenémicos y, en el caso del tratamiento dirigido, en aislamientos resistentes a los antibióticos de elección, principalmente en infecciones graves.
- Pacientes de bajo riesgo:
Una revisión sistemática no demostró diferencias significativas en el fracaso del tratamiento ni en la mortalidad asociada a infecciones en los pacientes de bajo riesgo tratados con antibioterapia vía oral de forma empírica frente a los que recibieron tratamiento intravenoso. Se han propuesto diferentes opciones (por ejemplo: levofloxacino, amoxicilina-clavulánico junto a ciprofloxacino, etc.), que habrá que valorar según la epidemiología local o infecciones previas del paciente.
Es importante realizar un seguimiento clínico estrecho del paciente. Si presentan deterioro clínico, intolerancia oral, signos de focalidad infecciosa de alto riesgo de progresión, o en los que se producen aislamientos microbiológicos sin opciones vía oral o en los que se prioriza la vía intravenosa, deberán ser ingresados. En los centros con posibilidad de tratamiento antimicrobiano domiciliario intravenoso, puede plantearse su utilización.
- Evaluación posterior de la neutropenia febril:
El tiempo medio de positividad de los hemocultivos en pacientes febriles neutropénicos es de unas 12h, siendo la mayoría positivos en las primeras 24h. Un crecimiento más allá de las 24-48h corresponde generalmente a microorganismos de crecimiento lento o microorganismos contaminantes.
Las últimas recomendaciones europeas en pacientes adultos emitidas por The European Conference on Infections in Leukemia (ECIL-4) establecen que en pacientes con NF sin documentación clínica ni microbiológica de infección, el tratamiento antibiótico empírico puede suspenderse después de al menos 72h de tratamiento intravenoso en pacientes que han estado hemodinámicamente estables desde la presentación y afebriles durante al menos 48h, independientemente del recuento de neutrófilos o de la duración esperada de la neutropenia.
Una guía internacional pediátrica considera segura la interrupción de la antibioterapia empírica en pacientes de bajo riesgo sin infección documentada, tras 24 horas de apirexia y 72h de antibioterapia, con independencia de la cifra de neutrófilos, sin establecer ninguna recomendación en el caso de persistir neutropenia grave.
- Tratamiento no antimicrobiano:
El tratamiento de soporte engloba medidas generales, como fluidoterapia, analgesia, antipiréticos, tratamientos habituales del paciente, valorar la necesidad de hidrocortisona, así como medidas dirigidas a disminuir la incidencia de infecciones en el paciente neutropénico. El tratamiento con el factor estimulador de colonias granulocíticas (G-CSF) acorta el tiempo de neutropenia, reduciendo el tiempo de antibioterapia e ingreso hospitalario, pero no ha demostrado disminuir las infecciones ni la gravedad de estas.
3.6.3 Trombosis en pediatría
La trombosis es una entidad infrecuente en la población pediátrica. A diferencia de los adultos, suele aparecer en niños con alguna patología de base, siendo rara la forma idiopática.
- Factores de riesgo para trombosis en niños:
- Catéter venoso central
- Cáncer
- Enfermedades cardiacas congénitas
- Trombofilias hereditarias
- Enfermedades autoinmunes
- Enfermedad renal (síndrome nefrótico)
- Deshidratación
- Sepsis
- Traumatismo / cirugía / inmovilización
- Obesidad
- Anticoncepción oral / tabaco (adolescentes)
- Asfixia perinatal / diabetes materna
- Enfermedad inflamatoria intestinal
- La clínica variará en función de la localización de la trombosis:
- Trombosis relacionada con catéter venoso central: generalmente asintomática, con disfunción del mismo.
- Trombosis venosa profunda (TVP) de extremidades: edema de una extremidad respecto a la contralateral, con dolor, eritema e incluso cianosis, palpación de cordón flebítico, ingurgitación venosa (dolor abdominal o inguinal) y, en casos avanzados, asimetría/pérdida de pulso. periférico.
- Trombosis de vena cava inferior: circulación colateral y disfunción hepática o renal.
- Trombosis de vena cava superior: cianosis y edema de cara, cuello y parte superior del tórax y, en estados avanzados, insuficiencia cardíaca congestiva.
- Trombosis de vena renal: hematuria, trombocitopenia, nefromegalia y oliguria.
- Trombosis portal: asintomática o hipertensión portal.
- Tromboembolismo pulmonar (TEP): aparición súbita de disnea, hipoxemia, taquicardia, dolor torácico, sibilancias, hemoptisis, síncope, hipertensión pulmonar e insuficiencia cardíaca.
- Trombosis arterial: en extremidades provoca mala perfusión y disminución de pulsos, en arteria renal aparece hipertensión arterial con o sin insuficiencia renal y en arteria mesentérica una enterocolitis necrotizante.
La principal complicación de la anticoagulación con heparina no fraccionada/sódica (HNF) o heparina de bajo peso molecular (HBPM) o anticoagulantes orales (warfarina o acenocumarol) es el sangrado.
3.6.4 Síndrome de vena cava superior
En los niños, la tráquea y los bronquios principales son muy vulnerables a la compresión, debido a que presentan diámetros intraluminales menores que los de los adultos. Hablamos de compresión traqueo bronquial cuando es producida por una masa mediastínica.
En la infancia, está casi siempre asociada a una obstrucción de la vena cava superior (también vulnerable a la compresión por su delgada pared y su baja presión intraluminal), produciendo el denominado síndrome de vena cava superior. Dichas alteraciones producen una elevación de la presión venosa y la dilatación retrógrada de las venas proximales a la obstrucción en cabeza, cuello, extremidades superiores y parte superior del tronco, promoviendo la aparición de colaterales. La suma de la compresión traqueobronquial y de la cava superior se denomina síndrome de mediastino superior y suele producirse por masas en mediastino anterior.
La mayoría de los niños y adolescentes que tienen una masa mediastínica van a presentar etiología maligna y, en la gran parte de los casos, se tratará de un linfoma, sobre todo, linfoblástico. También, es relativamente frecuente en las leucemias linfoblásticas de estirpe T. El incremento en el uso de dispositivos centrales, ha aumentado la prevalencia de síndrome de vena cava superior relacionado con trombosis. La compresión, además, puede ser debida a adenopatías.
La clínica varía según el grado de obstrucción, su localización y la velocidad de instalación. Los pacientes pueden estar asintomáticos en el momento del diagnóstico o presentar síntomas similares a los de una infección respiratoria, como tos, fiebre o sibilancias. Las manifestaciones iniciales también pueden incluir edema facial, venas prominentes superficiales en el tórax y en el cuello, ortopnea y estridor. Otros síntomas que pueden aparecer son plétora o cianosis facial, en el cuello o en los miembros superiores; cefalea que aumenta con el decúbito; quemosis; edema periorbitario; síndrome de Bernard Horner; disfagia y disfonía; y vértigo o acufenos. Comparado con los niños mayores y los adultos, los niños pequeños tienen menor probabilidad de presentación de síncope, confusión o alteraciones de la visión. La gravedad de los síntomas no siempre se correlaciona con el grado de compromiso de la vía aérea.
El compromiso hemodinámico y respiratorio pueden empeorar con la posición en supino o en flexión (al realizar una punción lumbar), con el ejercicio físico o el estrés emocional.
El método de diagnóstico inicial de elección es la radiografía simple de tórax. En ella se puede apreciar ensanchamiento mediastínico, así como signos indirectos de obstrucción (desviación de la tráquea o reducción del calibre de la vía aérea). También puede haber derrame pleural y pericárdico asociado.
Otra prueba diagnóstica importante es la tomografía computarizada (TAC), teniendo en cuenta factores de riesgo, como la posición en supino o la necesidad de sedación. Es la técnica que mejor permite estimar el grado de compresión de la tráquea y de la vena cava superior. Además, permite describir las principales características de la masa, tener una primera aproximación etiológica y aportar información sobre el estadiaje.
En la mayoría de los casos, se establece de forma gradual, por lo que siempre que sea posible hay que intentar realizar un diagnóstico etiológico antes de iniciar el tratamiento. Es frecuente que la respuesta al tratamiento dirigido sea rápida y, por ello, es la medida de elección siempre que el paciente se encuentre estable. Para el diagnóstico definitivo, deben emplearse las pruebas menos invasivas posibles.
En caso de que sea necesario el uso de pruebas que precisan sedación o anestesia general, debe ser evaluado el riesgo de mayor compromiso de vía aérea con dichas actuaciones. Las complicaciones asociadas a la anestesia son más frecuentes si el paciente presenta: ortopnea, edema del hemicuerpo superior, disnea en reposo, disminución del diámetro traqueal mayor del 50% respecto a lo normal para la edad y sexo del paciente, compresión grave de uno o ambos bronquios principales, pico espiratorio de flujo menor del 50% o derrame pericárdico asociado. La ortopnea es el parámetro que más se correlaciona con el riesgo. Si es necesaria intubación para algún procedimiento, debe ser valorado de forma multidisciplinar el riesgo ya que puede ser difícil la extubación posterior.
Entre las medidas terapéuticas generales se encuentran: posición elevada de cabeza y cuello aproximadamente 30-45º, mantener situación de calma en el niño, oxígeno suplementario y, en ocasiones, puede ser necesario el uso de diuréticos. Hay que tener precaución con la hiperhidratación, vigilando equilibrios de forma estrecha. En los casos relacionados con trombosis del catéter central, puede estar indicado su retirada, así como la anticoagulación sistémica y, en algunas ocasiones, la implantación de un stent.
Si estamos ante un caso grave, con compromiso vital, puede ser necesario iniciar el tratamiento antes de tener un diagnóstico etiológico, valorando el uso de corticoides, agentes quimioterápicos (la ciclofosfamida) e incluso la radioterapia local. Dichas medidas pueden dificultar el diagnóstico histológico posterior, aunque se apliquen durante poco tiempo.
3.6.5 Cistitis hemorrágica
Se define como la presencia de hematuria mantenida asociada a síntomas del tracto urinario inferior, en ausencia de infección urinaria (ITU) bacteriana o fúngica. Se trata de una cistitis estéril no infecciosa asociada a hematuria franca.
Ocurre, principalmente, en pacientes tratados con ifosfamida y altas dosis de ciclofosfamida, así como en niños sometidos a trasplante de progenitores hematopoyéticos (TPH).
Está causado por:
- Varios agentes quimioterápicos: busulfán, doxorrubicina, fludarabina y, sobre todo, ifosfamida y ciclofosfamida.
- Radiación pélvica.
- En el contexto del trasplante hematopoyético, por infecciones víricas.
La presentación clínica es muy variable; desde hematuria microscópica con disuria, polaquiuria y urgencia miccional escasas, a hematuria masiva con intenso dolor.
En los casos de hematuria secundaria a quimioterapia, esto suele aparecer a las 24-48 horas de la administración. Sin embargo, en el caso de la radioterapia, su aparición puede retrasar meses o años desde la administración del tratamiento.
El diagnóstico se basa en una clínica compatible con el antecedente de exposición a quimioterapia, radioterapia y/o TPH (trasplante de progenitores hematopoyéticos), descartando otras causas potenciales de esta sintomatología (ITU bacteriana o fúngica, urolitiasis, invasión tumoral). Entre las pruebas necesarias para el diagnóstico, se incluyen:
- Sedimento y urinocultivo.
- Otras pruebas de imagen en función de la gravedad del cuadro: ecografía de abdomen, TAC, resonancia magnética nuclear (RMN), cistografía o cistoscopia.
- En pacientes sometidos a TPH, detección de virus en sangre u orina.
El mejor tratamiento de la cistitis hemorrágica es una profilaxis adecuada. En todos los pacientes que reciben ifosfamida o ciclofosfamida, debe realizarse prevención mediante hiperhidratación (3 l/m2) y administración de 2-mercapto-etano-sulfonato de sodio (MESNA), que protege la mucosa vesical. Además, se mantendrá una estrecha vigilancia durante el tratamiento, con controles de tiras de orina.
En caso de obstrucción del flujo urinario por la presencia de coágulos, el siguiente paso es la colocación de un sondaje vesical para realizar un lavado vesical continuo con suero salino. Debe administrarse de forma concomitante una analgesia correcta, así como profilaxis antibiótica.
Si las medidas previas no son suficientes, se realizará una cistoscopia que permite localizar puntos sangrantes y extracción de coágulos.
3.6.6 Náuseas y vómitos
A pesar del uso generalizado de pautas de tratamiento altamente eficaces en la prevención de las náuseas y vómitos inducidos por quimioterapia, este efecto adverso continúa presentándose en un porcentaje importante de pacientes, dependiendo del régimen quimioterápico utilizado y de las características del paciente.
Las náuseas y vómitos continúan estando entre los efectos adversos de la quimioterapia que más ansiedad provocan en los pacientes, teniendo una gran repercusión sobre la calidad de vida y obligando, en algunos casos, a posponer, cambiar o suspender los tratamientos, debido a la aparición de complicaciones, como deshidratación, desequilibrio electrolítico, etc.
Los fármacos quimioterápicos se agrupan en 4 niveles de intensidad, en función de la probabilidad que tienen de producir náuseas o vómitos si no reciben un tratamiento adecuado para evitarlos o prevenirlos. Estos niveles nos orientan sobre el tratamiento antiemético que se debe administrar.
- Riesgo alto: más del 90% de incidencia de náuseas y vómitos si no administramos un tratamiento antiemético. Por ejemplo, en los esquemas con cisplatino.
- Riesgo moderado: náuseas y vómitos en el 30-90% de los pacientes. Carboplatino, Oxaliplatino.
- Riesgo bajo: Náuseas y vómitos en el 10-30% de los pacientes. Paclitaxel, Docetaxel, Etoposido, Topotecán, Pemetrexed, Gemcitabina.
- Riesgo mínimo: Menos del 10% de los pacientes presentarán náuseas y/o vómitos si no reciben un tratamiento adecuado. Bevacizumab, Erlotinib, Gefitinib, Cetuximab, Vinorelbina.
Además, hay que tener en cuenta que la mayoría de los tratamientos oncológicos son esquemas que combinan varios fármacos quimioterápicos, por lo que el riesgo de emesis se ve incrementado.
3.6.7 Mucositis
La mucositis se produce como consecuencia de la lesión de las células epiteliales y mucosas que recubren todo el tracto gastrointestinal. Las alteraciones pueden variar desde el simple eritema hasta la ulceración y/o necrosis grave. Los principales responsables son la quimioterapia y la radioterapia utilizadas para el tratamiento de la enfermedad tumoral. Otros determinantes que pueden empeorar la afectación son: presencia de caries, patología periodontal, neutropenia o mala higiene bucal.
En la mucosa oral, puede producir eritema y/o edema, sensación de quemazón, dolor, sequedad o alteraciones del gusto, ulceración, sangrado y dificultad en el habla o para abrir la boca. En los casos más graves, puede aparecer una membrana mucosa blanquecina debido a úlceras confluentes. Las manifestaciones en caso de afectación gastrointestinal pueden ser dolor abdominal difuso o diarrea acuosa con dolor, y en casos graves, pueden complicarse con perforación, formación de fístulas y obstrucción intestinal.
Existen numerosas escalas para determinar la gravedad de la afectación oral. Una de las más utilizadas es la de la Organización Mundial de la Salud (OMS):

La mucositis predispone a la aparición de infecciones secundarias, ya que los pacientes normalmente presentan neutropenia asociada, destacando la candidiasis orofaríngea y la estomatitis herpética.
El tratamiento de la mucositis es básicamente de soporte, siendo importante:
- Mantener una higiene bucal adecuada, evitando soluciones alcohólicas que reseca la mucosa y que pueden empeorar el dolor.
- Analgesia adecuada: desde frío local o anestésicos tópicos, como la lidocaína viscosa al 2%. En ocasiones, es necesario el empleo de opiáceos.
- Tratamiento tópico con ácido hialurónico.
- En caso de sobreinfección fúngica y/o por virus herpes simple, instaurar tratamiento específico.
- Alimentación blanda y fría, evitando ácidos. Valorar de forma estrecha la capacidad de ingesta y el estado nutricional del paciente para administrar suplementos en casos necesarios e incluso instaurar nutrición parenteral en casos graves y prolongados.
- Vigilar la frecuencia y características de las deposiciones, así como la presencia de sangrado o mucosidad que puede indicar afectación grave del tracto digestivo inferior.
3.6.8 Tiflitis
También denominado síndrome ileocecal o enterocolitis necrotizante, debido a que el ciego está casi siempre afectado por su distensibilidad y flujo sanguíneo limitado. Sin embargo, el término más correcto es el de enterocolitis neutropénica, que hace referencia a su patogenia. Puede afectar a cualquier segmento del tracto gastrointestinal y es debida a invasión bacteriana y/o fúngica de la pared abdominal, siendo frecuente la etiología polimicrobiana.
El factor de riesgo más importante es la administración de altas dosis de citostáticos, especialmente citarabina y etopósido. El estreñimiento y el íleo favorecen el crecimiento bacteriano y fúngico en la luz intestinal, aumentando también el riesgo de infección, especialmente si existe mucositis asociada. También, puede intervenir en su patogenia la infiltración de la pared por células neoplásicas. Se describe con más frecuencia en pacientes con leucemia aguda, sobre todo, mieloblástica y linfoblástica en inducción, y en linfomas de Burkitt, siendo menor en tumores sólidos.
La incidencia está en aumento debido al uso de regímenes de quimioterapia más intensivos, el aumento de la supervivencia y la optimización del diagnóstico.
La clínica suele aparecer a los 10-14 días después de finalizar la quimioterapia. Se debe sospechar el diagnóstico ante un paciente neutropénico con dolor abdominal, sobre todo, en cuadrante inferior derecho o difusa, distensión abdominal y fiebre. También puede presentar: náuseas, vómitos, diarrea, a menudo acuoso y con hebras sanguinolentas, y disminución de los ruidos intestinales. A veces, a la palpación impresión de masa en fosa ilíaca derecha. Dado que los síntomas son bastante inespecíficos, el diagnóstico puede resultar difícil y, a menudo, no se hace de forma temprana. Así mismo, hay que tener en cuenta que el tratamiento con corticoides puede enmascarar los signos de inflamación.
Las técnicas de imagen de elección son la ecografía y la TAC abdominal, en las que se aprecia engrosamiento mural intestinal (mayor de 3-5 mm en transverso según los distintos autores. No se recomienda la realización de endoscopia digestiva, por el riesgo de perforación.
Se trata de una situación grave que puede complicarse con sepsis, shock séptico, coagulación intravascular diseminada (CID), oclusión, perforación intestinal, peritonitis y hemorragia digestiva. Se describe una mortalidad de hasta el 11%, por lo que, en un primer momento, es importante valorar la presencia de signos de gravedad.
El tratamiento es conservador en la mayoría de los pacientes. Requiere: reposo intestinal, hidratación, soporte transfusional, oxigenoterapia en casos necesarios, administración de factor estimulante de colonias de granulocitos (G-CSF), valoración de sonda nasogástrica, suspensión de la medicación antitumoral y analgesia adecuada, evitando derivados opiáceos en la medida de lo posible, dado que favorecen el íleo paralítico. Cuando el reposo digestivo se prolongue, se iniciará nutrición parenteral. También es necesario comenzar de forma precoz antibióticos de amplio espectro, recomendándose meropenem de primera elección (cefepime + metronidazol como alternativa) y, en los casos graves, asociar amikacina, vancomicina, +/- metronidazol y +/- antifúngicos. Es importante descartar infección concomitante por Clostridium difficile. En pacientes con afectación leve-moderada, la normalización de la cifra de neutrófilos logra generalmente contener el proceso.
Ante la sospecha de complicaciones, será preciso la valoración por Cirugía Pediátrica, especialmente en caso de peritonitis, perforación intestinal, hemorragia persistente pese a corrección de coagulopatía y trombopenia, así como deterioro clínico que no responde al tratamiento médico.
3.6.9 Dolor
Los pacientes con cáncer a menudo deben someterse a procedimientos invasivos, de larga duración, hospitalizaciones de manera recurrente y se enfrentan a cuidados en casa que alteran el funcionamiento habitual de los roles en el hogar. Durante la fase diagnóstica se presentan episodios de dolor que se mantienen a lo largo del tratamiento, especialmente en los periodos de hospitalización, lo que hace necesario el manejo de este síntoma para lograr una mejor adaptación a la situación. Por tanto, con una adecuada identificación de la experiencia dolorosa se puede elegir el tratamiento más apropiado para cada caso.
3.6.10 Síndrome de lisis tumoral
El término síndrome de lisis tumoral hace referencia al conjunto de alteraciones metabólicas resultantes de la rápida destrucción de las células malignas y cuyas consecuencias pueden ser potencialmente graves. Se caracteriza por la presencia de hiperuricemia, hiperkaliemia, hiperfosfatemia e hipocalcemia, las cuales pueden aparecer de manera espontánea o en los primeros días tras el inicio del tratamiento citotóxico.
Las alteraciones de laboratorio surgen de la liberación masiva de metabolitos intracelulares cuyo catabolismo da lugar a la producción de ácido úrico. El exceso de estos productos supera la capacidad excretora del riñón, lo que lleva a su acumulación y posterior desarrollo del síndrome de lisis tumoral. La producción excesiva de ácido úrico favorece la precipitación del mismo en forma de cristales en los túbulos renales; de igual manera, la hiperfosfatemia favorece la formación y precipitación de cristales de fosfato cálcico, dando lugar a la aparición secundaria de hipocalcemia. Consecuencia de lo anterior, se puede producir una nefropatía obstructiva que desemboca en una insuficiencia renal aguda. El daño renal agudo favorece el desarrollo del síndrome de lisis tumoral, al dificultar la eliminación de los metabolitos, al mismo tiempo que origina el daño renal.
MANIFESTACIONES CLÍNICAS:
Las manifestaciones clínicas incluyen náuseas, vómitos, calambres, tetania, letargia, convulsiones e incluso riesgo de arritmias y parada cardíaca.
El síndrome de lisis tumoral ocurre con mayor frecuencia en pacientes afectados de linfomas no Hodgkin (LNH), sobre todo, los de tipo Burkitt, leucemias agudas linfoblásticas (LLA), especialmente las de estirpe T y leucemias agudas mieloblásticas (LMA).
PROFILAXIS:
La profilaxis del síndrome de lisis tumoral se basa en:
- Líquidos: se recomienda instalar hiperhidratación. Para mejorar la excreción urinaria, puede ser necesario el empleo de diuréticos, como furosemida (0,5-1 mg/kg), salvo en situaciones de uropatía obstructiva o hipovolemia en las que su uso estaría contraindicado. El objetivo de estas medidas es aumentar el volumen intravascular, el flujo renal y el filtrado glomerular, favoreciendo así la excreción de fósforo y ácido úrico.
- Agentes uricosúricos: en la prevención del síndrome de lisis tumoral se emplean:
- Alopurinol: es un inhibidor de la enzima xantina oxidasa, que actúa bloqueando la conversión de hipoxantina en xantina y de esta en ácido úrico. Disminuye la formación de ácido úrico y la precipitación de cristales a nivel de los túbulos renales, pero no elimina el previamente formado, por lo que su efecto terapéutico se retrasa entre 24-72 horas. Se debe iniciar entre 12 y 24 horas antes del inicio de la quimioterapia.
- Rasburicasa: forma recombinante de la enzima urato oxidasa, que transforma el ácido úrico en alantoína (5-10 veces más soluble que el ácido úrico). Inicio de acción inmediata, actuando sobre al ácido úrico preexistente y disminuyendo los niveles en las primeras 4 horas tras su administración, lo que permite iniciar el tratamiento quimioterápico de forma más segura respecto al empleo de alopurinol. Se administra por vía intravenosa.
- Monitorización estrecha: control de diuresis, recurriendo al sondaje vesical si fuera preciso, con balance de líquidos cada 8 horas. Control analítico que incluye: hemograma, perfil renal, ácido úrico, iones y gasometrías frecuentres.
TRATAMIENTO:
El objetivo del tratamiento es corregir las alteraciones metabólicas existentes y evitar la aparición de un daño renal agudo. Es fundamental el manejo de líquidos, asegurando una diuresis adecuada mediante la estrecha monitorización de los pacientes.
- Hiperuricemia: hiperhidratación y rasburicasa. Se recomienda mantener el tratamiento durante 3-7 días, en función de los niveles de ácido úrico. Las muestras de sangre deben colocarse en hielo, para evitar la degradación del ácido úrico por la rasburicasa a temperatura ambiente y asegurar un adecuado control de los niveles.
- Hiperfosfatemia: en los casos asintomáticos, se debe asegurar una adecuada hidratación y diuresis. Ocasionalmente, se han empleado quelantes del fósforo, como el hidróxido de aluminio, pero su inicio de acción lento, mala tolerancia y la toxicidad acumulada por el aluminio hacen que no se recomiende de forma rutinaria.
- Hipocalcemia: Los casos asintomáticos no requieren tratamiento, ya que la elevación de los niveles puede favorecer la formación y precipitación de cristales de fosfato-cálcico. En los pacientes sintomáticos, se debe instaurar tratamiento con gluconato cálcico al 10% y monitorización electrocardiográfica.
- Hiperkaliemia: En pacientes asintomáticos con K ≥6 mEq/L, se debe evitar el aporte exógeno de potasio, administrar furosemida intravenosa e iniciar tratamiento con resinas de intercambio iónico. Por el contrario, en pacientes sintomáticos y/o K ≥ 7 mEq/L, se deben iniciar medidas de manera urgente:
- Gluconato cálcico al 10% (1 ml/kg ev en 10 minutos). Es necesario realizar monitorización electrocardiográfica y, en caso de bradicardia, suspender.
- Bicarbonato sódico 1M (1-2 mEq/kg incluso en 10-20 minutos).
- Insulina regular (0,1-0,2 UI/kg) en suero glucosado (0,5-1 g/kg).
- Furosemida intravenosa (1 mg/kg).
- Salbutamol nebulizado (0,15 mg/kg) o intravenoso (5 mcg/kg en 15 minutos).
Cuando estas medidas fracasan y se producen un daño renal agudo hay que recurrir a técnicas de depuración extrarrenal.
3.6.11 Síndrome de secreción inadecuada de hormona antidiurética (SIADH)
Dentro de las entidades que cursan con hiponatremia, cabe destacar por su frecuencia, hasta un 30% de las hiponatremias en niños oncológicos, el síndrome de secreción inadecuada de hormona antidiurética (SIADH).
La hiponatremia es una de las alteraciones electrolíticas más frecuentes en los pacientes con cáncer. Se define por la presencia de cifras de sodio sérico (Na)<135 mEq/l, considerándose hiponatremia grave cifras <125 mEq/l, a partir de las cuales el riesgo de edema cerebral aumenta significativamente. La rapidez de instalación de la hiponatremia es determinante en la presentación clínica, siendo los síntomas neurológicos, como somnolencia, letargia, convulsiones o coma, los más llamativos.

Surge como resultado de una secreción excesiva de hormona antidiurética (ADH), que provoca un aumento en la reabsorción de agua en los túbulos, con la consiguiente hemodilución que da lugar a la hiponatremia. Para intentar compensar, se produce un aumento de la natriuresis, pero debido a la situación de antidiuresis la orina persiste hipertónica y la hiponatremia se mantiene.
La causa más frecuente en los pacientes pediátricos son los agentes quimioterápicos, entre los que destacan vincristina, vinblastina, los agentes alquilantes, cisplatino y melfalán. Otros factores etiológicos serían el daño directo sobre el tejido cerebral, tanto por cirugía como radioterapia.
Dentro del tratamiento, en los casos sintomáticos, se incluye la restricción hídrica (50% de las necesidades basales), siendo esta especialmente compleja en aquellos pacientes en tratamiento con agentes quimioterápicos que precisan aumento de los aportes de líquidos para su administración. El empleo de diuréticos, como la furosemida, puede ser de utilidad en los casos graves para favorecer la eliminación del agua libre. Es importante mantener aportes adecuados de sodio, realizando correcciones hasta 130 mEq/L con ClNa 3%.
3.6.12 Enfermedad venooclusiva hepática (EVOCH)
El síndrome caracterizado por ictericia, hepatomegalia y aumento de peso por retención hídrica, desarrollado de forma precoz tras un trasplante de progenitores hematopoyéticos (TPH), suele denominarse enfermedad venooclusiva hepática (EVOCH), si bien lo correcto sería emplear la nueva terminología de síndrome de obstrucción sinusoidal. Desde la primera descripción en 1979, el TPH se ha convertido en la principal causa de EVOCH y ésta en una de las principales causas de morbilidad y mortalidad tras el TPH.
El 75-85% de los casos de EVOCH se resuelven a los 15-25 días de su inicio, pero el 15-25% de los pacientes pueden fallar como consecuencia directa de la venooclusión o de complicaciones relacionadas.
Actualmente, se piensa que el agente con mayor capacidad preventiva probablemente sea el que ha mostrado tener la mayor efectividad terapéutica, el defibrótido. La experiencia con este agente queda limitada a unos pocos estudios con un total de 124 pacientes en los que se observó una clara reducción en la incidencia de EVOCH, y un amplio estudio aleatorio realizado en población pediátrica que puso de manifiesto una reducción significativa de la misma, así como un impacto aún mayor en la incidencia de enfermedad injerto contra huésped. Esta observación, y el resultado de estudios in vitro, ponen de manifiesto que este agente tiene un potente efecto modulador sobre la célula endotelial, efecto que podría justificar estos resultados.
3.6.13 Catéter Venoso Central: Reservorio subcutáneo
Un reservorio venoso subcutáneo, conocido como porth-a-cath, es un catéter venoso central de larga duración que consta de un reservorio o puerto subcutáneo generalmente de titanio, con una membrana de silicona conectada a un catéter de silicona radiopaca o poliuretano. Su porción proximal está tunelizada, es introducido por la vena subclavia o yugular y llega a la vena cava superior.
Fuente: Dumont C. Posición de un reservorio implantado. Nurse Key. Infusion Therapy. 2017.
Se trata de un catéter interno insertado con técnica tunelizada y totalmente implantado a nivel subcutáneo en el tórax, permitiendo el acceso repetido al sistema vascular, facilitando tanto la extracción de muestras de sangre como la administración de fármacos, que cuenta con el reservorio subcutáneo de titanio con membrana de silicona mencionada anteriormente (queda ubicado preferentemente en el hemitórax derecho) no tiene partes externas visibles y se accede a través de una punción en la piel con técnica estéril de una aguja tipo Huber® o Gripper® que no daña la silicona del portal.
Fuente: Dumont C. Aguja para acceder a reservorio. Nurse Key. Infusion Therapy. 2017.
- Los profesionales implicados para realizar la punción del reservorio son:
- Personal facultativo o enfermeros entrenados en la técnica de la manipulación de catéteres venosos centrales.
- Técnicos de cuidados auxiliares de enfermería (TCAE): encargados de la preparación del material, colaboran durante la realización del procedimiento e intervienen en la colocación y sujeción del paciente pediátrico en caso necesario.
- Celador: encargados de la colocación y sujeción del paciente pediátrico en caso necesario.
- Desarrollo del procedimiento:
- Información paciente/familia:
Información de la técnica a realizar en un ambiente seguro y tranquilo.
Comprobar la identidad del paciente verificando la pulsera identificativa del mismo y confirmando esa identidad con los padres/tutores o cuidadores presentes en el momento de realización de la técnica.
Es importante preparar al paciente y a sus padres/cuidadores para el procedimiento que se le va a realizar mediante explicaciones claras, apropiadas y adaptadas a la edad y/o el estado cognitivo.
La información que se les debe proporcionar debe contener los objetivos del procedimiento y los resultados esperados tras su realización. Para facilitar su compresión, se pueden utilizar muñecos, dibujos o vídeos adaptadores si se encuentran disponibles en su unidad. Los objetivos son: comprobar que el niño comprende el procedimiento, disminuir el miedo y la ansiedad.
- Preparación del entorno:
Intente crear un ambiente tranquilo, con control de la intensidad luminosa, del ruido y de la temperatura de la sala.
Inicie medidas de distracción apropiadas a la edad del niño y a su situación clínica y asegúrese de que se mantiene en la medida de lo posible mientras dure el procedimiento.
Los padres deben, en la medida de lo posible, estar presentes para ayudar a los niños a afrontar el procedimiento. Para ello proporcione instrucciones claras sobre el papel que se espera que realicen durante la realización del procedimiento. Estas instrucciones deben estar enfocadas a tranquilizar al niño y disminuir el estrés durante la realización del procedimiento.
Debe preservarse en todo momento la intimidad del paciente.
- Preparación de material: La preparación del material que se va a utilizar debe realizarse fuera de la vista del niño para disminuir la ansiedad anticipatoria. En un lugar limpio y accesible debe colocarse todo el equipo a utilizar que debe constar de:
- Paño y gasas estériles.
- Guantes limpios y estériles.
- Mascarilla quirúrgica.
- Esponja jabonosa.
- Solución hidroalcohólica para limpieza de manos.
- Agujas tipo Huber® o Gripper® de 20 o 22 G.
- Jeringas estériles de 10 ml.
- Antisépticos: preferentemente clorhexidina alcohólica al 2 % (piel intacta) o, en caso de alergia, alcohol al 70 %. Clorhexidina acuosa al 2% (piel con lesión o tras cambio de aguja).
- Llave de tres pasos y bioconector o tapón antirreflujo.
- Tubos estériles para toma de muestra sanguínea.
- Campana de extracción o sistema Vacutainer® (En caso de necesidad de extracción de analítica sanguínea).
- Suero salino fisiológico (SSF) al 0,9 %.
- Heparina: Solución heparinizada o fórmulasfabricadas de heparina sódica vial de 20 UI/ml.
- Material necesario para realizar el procedimiento requerido: solución de hemoderivados, sueroterapia, sistemas de perfusión, fármacos, etc.
- Material de fijación: Apósito de gasa estéril o transparente semipermeable estéril.
- Realización de la técnica:
Colocar al paciente en la camilla en decúbito supino y girar su cabeza hacia el lado contario a la localización del reservorio.
Limpieza de la zona de inserción (esponja jabonosa + suero fisiológico + gasas estériles para secado).
Asepsia de la zona de inserción: gire la cabeza al lado contrario del punto de inserción y aplique clorhexidina alcohólica al 2% y deje actuar 30 segundos.
Preparación del campo y material estéril.
Colocarse el equipo apropiado (guantes estériles y mascarilla quirúrgica) y lavarse las manos con agua y jabón o solución hidroalcohólica.
Colocar el paño estéril alrededor o por debajo de la zona de punción.
Purgar el sistema de pinza de aguja con suero fisiológico de forma estéril y sujetarlo.
Localizar sitio de la punción.
Aplique un antiséptico local en forma circular, desde el punto de inserción hacia el exterior cubriendo un área aproximada de 10cm. Usando una única gasa en una sola dirección y dejar actuar durante 30-60 segundos.
Localizar el catéter venoso central: Localizarlo por palpación con la mano no dominante y sujetar sin hacer demasiada fuerza ni estirar. Pedir al paciente que inspire e introduzca la aguja tipo Huber® o Gripper® (previamente purgada) en ángulo de 90º con la mano dominante, hasta notar una base dura metálica que es la base del reservorio.
Conectar la jeringa de 10 ml y aspirar para comprobar la correcta colocación de la aguja y la permeabilidad de la vía mediante la extracción de 3-5 ml de sangre (así se retira la heparina del sellado anterior). Colocar tapón antirreflujo (previamente purgado con suero fisiológico). Evitar siempre la entrada de aire al sistema.
Conectar una nueva jeringa o campana de extracción y extraer el volumen de sangre requerido o, en su caso, proceder a la administración de fármacos, perfusiones o transfusión de hemoderivados.
El número de botes de hemocultivo y el volumen de sangre a introducir en ellos dependerán de lo establecido por cada centro, por lo que se deben seguir siempre las indicaciones de los protocolos internos. No obstante, por regla general, en pacientes menores de 12 kg se extraerá una botella de hemocultivo pediátrico de origen central y otra de origen periférico. El volumen a introducir en cada bote, de manera estéril y previa descontaminación de las siliconas con clorhexidina alcohólica, será de 1-2 ml de sangre/botella. En mayores de 12 kg se repetirá el procedimiento descrito anteriormente, pero introduciendo 5 ml/botella.
Posterior al uso del catéter, irrigar el sistema con una nueva jeringa con 10 ml de suero fisiológico. Se realizará mediante la técnica de lavado en “pulsos” (inyectar 1 ml, parar y volver a inyectar), que produce turbulencia dentro de la luz del catéter y favorece la eliminación de los restos de fibrina.
Sellado del catéter: Sellar el catéter con 3 ml de solución heparinizada o fórmulas fabricadas de heparina sódica 20 UI/ml o suero fisiológico. Algunos estudios no han encontrado diferencias significativas entre ambos métodos en términos de prevención de obstrucciones. Introducir el suero fisiológico o la solución heparinizada realizando una presión positiva; es decir, sin dejar de ejercer presión en el embolo de la jeringa, clampar con la mano no dominante cuando queden aproximadamente 0,5-1 ml, para evitar el reflujo en la punta del catéter.
Fijación de la aguja: Si se va a seguir utilizando el reservorio, no hay que retirar la aguja, si no que debemos fijar la aguja a la piel, colocando un apósito para cubrirla.
Si el catéter se va a utilizar solo para extracción sanguínea, se debe extraer la aguja posterior al sellado del sistema. Para ello, estabilizar el reservorio con dos dedos de la mano no dominante, pedir al paciente que haga una inspiración forzada y retirar la aguja con un tirón suave, pero firme. Colocar un apósito estéril con gasa o una gasa con solución antiséptica y esparadrapo sobre el punto de inserción, manteniendo una presión durante dos minutos.
- Complicaciones/problemas potenciales:
- Extravasación
- Necrosis cutánea
- Bacteriemia relacionada con el catéter
- Trombosis venosa
- Embolismo aéreo
- Oclusión parcial o total (fibrinolítico: urokinasa).
- Registro del procedimiento:
Documente el procedimiento en la hoja de cuidados o en el registro electrónico de enfermería de cada unidad o servicio hospitalario según los protocolos establecidos para ello. Las listas de verificación tipo CHECK-LIST son muy útiles para no olvidar nada del procedimiento.
BIBLIOGRAFÍA
- Almaguer YC, Pupo AS, Criollo LMT. Atención al paciente politraumatizado pediátrico. 2021.
- Anatomy of the Endocrine System in Children. Available at: https://www.stanfordchildrens.org/es/topic/default?id=anatomy-of-the-endocrine-system-in-children-90-P05044. Accessed Jun 27, 2024.
- Arroyo Díez FJ. Lo que debes saber sobre la diabetes en la edad pediátrica (4a. 2019).
- Barrios Castellano, R. Diabetes tipo 1 en la edad pediátrica. Lo que se necesita saber para la autogestión de la diabetes (2021).
- Benito Bernal AI, Vila de Frutos R. Neuroblastoma y tumores relacionados. Pediatría integral 2021;25(7):340.e1–340.e16.
- Cañete Nieto A, Pardo Romaguera E, Alfonso Comos P, Valero Poveda S, Porta Cebolla S, Valderrama Zurián JC, et al. Cáncer infantil en España. Estadísticas 1980-2023.
Registro Español de Tumores Infantiles (RETI-SEHOP). Universitat de València 2024. - Carreras E. Prevención y tratamiento de la enfermedad venooclusiva hepática. Gastroenterología y Hepatología 2011;34(9):635–640.
- Cebrián Muíños C. Criptorquidia y patología testículo-escrotal en la edad pediátrica. Pediatría integral 2019;23(6):271–282.
- Cid JL. Manual de cuidados intensivos pediátricos. : Publimed; 2013.
- Corral Sánchez MD, Tarabini-Castellani Ciordia BM. Tumor de Wilms y otros tumores renales. Pediatría integral 2021;25(7):341–347.
- Drake RL, Vogl AW, Mitchell AM. Gray. Anatomía para estudiantes. : Elsevier Health Sciences; 2020.
- Díez YB. Manejo del paciente politraumatizado. 2020.
- Fernández-Delgado R. La oncología pediátrica: pasado, presente y futuro. Anales de Pediatría 2016;85(2):59–60.
- Fernández-Plaza S, Reques Llorente B. Bases del tratamiento del cáncer en Pediatría: principios de la terapia multimodal. Pediatría integral 2016;20(7):465–474.
- García-Salido A, La Calle GH, González AS. Revisión narrativa sobre humanización en cuidados intensivos pediátricos: ¿dónde estamos? Medicina Intensiva 2019;43(5):290–298.
- Garreta Aperte ME. Técnicas y procedimientos de Enfermería. 2018.
- Guerrero-Fernández J, Sánchez AJC, Bonis ACB, Suso JM, Domínguez JR. Manual de diagnóstico y terapéutica en Pediatría. ; 2018.
- Herramienta online para la consulta y diseño de Planes de Cuidados de Enfermería. NNNConsult. Elsevier. 2015.
- Hockenberry MJ, Rodgers CC. Wong. Enfermería Pediátrica. : Elsevier; 2019.
- Hospital Universitario Virgen del Rocío. Manual de atención inicial al paciente pediátrico con traumatismo grave.
- Herramienta online para la consulta y diseño de Planes de Cuidados de Enfermería. NNNConsult. Elsevier. 2015; Available at: https://www-nnnconsult-com.bvsspa.idm.oclc.org/nanda.
- Iniciativa mundial para el desarrollo de registros de cáncer. TNM Esencial Guía del usuario. International Agency for Research Cancer. World Health Organization 2022.
- López Laso E, Mateos González ME. Tumores cerebrales infantiles, semiología neurológica y diagnóstico. Protoc diagn ter pediatr 2022;1:151–158.
- López Fernández E, Oviedo Melgares L, Ordoñez Sáez O, Belda Hofheinz S, Ramos Casado MV. Viabilidad y aceptación de los diarios de la UCI adaptados al paciente pediátrico, ¿una nueva herramienta contra el síndrome poscuidados intensivos familiar? Anales de Pediatría 2023;98(4):308-310.
- Macías Franco S, Rozas Reyes P. Patología congénita ocular. Pediatría integral 2018;22(1):6–15.
- Martínez Campos L, Pérez-Albert P, Ferres Ramis L, Rincón-López EM, Mendoza-Palomar N, Soler-Palacin P, et al. Documento de consenso de manejo de neutropenia febril en el paciente pediátrico oncohematológico de la Sociedad Española de Infectología Pediátrica (SEIP) y la Sociedad Española de Hematología y Oncología Pediátrica (SEHOP). An Pediatr (Barc) 2023;98(6):446–459.
- Márquez-Díaz RR. Impacto de una intervención educativa sobre conocimientos del síndrome post-UCI pediátrico en cuidadores de niños hospitalizados. Proyecto de investigación. Nure Investigación 2023;20(124):1-8.
- Mendoza Sánchez MC, Riesco Riesco S, González Prieto A. Urgencias oncológicas en Pediatría. Pediatría integral 2019;23(2):65–80.
- Mezquita Romero L, Molinos Quintana A, Pérez Hurtado JM. Trombosis en la edad infantil. Manuales Clínicos 2022.
- Ministerio de Sanidad. Cuidados Paliativos Pediátricos en el Sistema Nacional de Salud: Criterios de Atención. 2014.
- Ministerio de Sanidad. Guía de Práctica Clínica sobre Cuidados Paliativos. 2008.
- Pérez Llarena G, Santos Ibáñez N. Punción port-a-cath. Sociedad Española de Urgencias de Pediatría (SEUP) 2021:1–8.
- Romo Muñoz MI, Núñez Cerezo V, Dore Reyes M, Vilanova Sánchez A, González-Peramato P, López Pereira P, et al. Tumores testiculares en la edad pediátrica: indicaciones de la cirugía conservadora. An Pediatr (Barc) 2018;88(5):253–258.
- Sastre Urgellés A, Rubio Aparicio P. Tumores óseos. Rabdomiosarcomas. Pediatría integral 2021;25(7):348–356.
- Suárez E, Serrano A. Atención inicial al traumatismo pediátrico. 2013;11(1):11-22.
- Tang M, Xu M, Su S, Huang X, Zhang S. Post-Intensive Care Syndrome in Children: A Concept Analysis. J Pediatr Nurs 2021;61:417-423.
- Traube C. Beware the Aftermath: Delirium and Post-Intensive Care Syndrome in Critically Ill Children. Pediatr Crit Care Med 2022;23(2):144-146.
- Toobe M. Síndrome post cuidados intensivos en pediatría. Rev Fac Cien Med Univ Nac Cordoba 2021;78(4):408-414.
- Vázquez Gómez F, Carceller Ortega E, Lassaletta Atienza A. Tumores cerebrales en niños. Pediatría integral 2021;25(7):357–366.
- Vázquez Rueda F, Murcia Pascual FJ, Siu Uribe A, Ortega Salas RM, Escassi Gil A, Garrido Pérez JI, et al. Análisis de los tumores sólidos ováricos pediátricos en nuestra población. An Pediatr (Barc) 2020;92(2):88–93.
- Wassner AJ. Thyroid nodules and cancer in children. UpToDate 2022.
- Wechsler DS. Overview of common presenting signs and symptoms of childhood cancer. UpToDate 2023.